|
x |
x |
|
 |
 |
|
I BỆNH NHIỄM TRÙNG |
VI KHUẨN HỌC |
MIỄN DỊCH HỌC |
NẤM HỌC |
KÝ SINH TRÙNG HỌC |
VIRÚT HỌC |
|
|
MIỄN DỊCH - CHƯƠNG MƯỜI
CHÍN
SUY GIẢM MIỄN DỊCH
Abdul
Ghaffar, Ph.D.
Emertius Professor of Pathology, Microbiology and Immunology
University of South Carolina
Biên dịch: Nguyễn Văn Đô, MD., PhD.,
Bộ môn Sinh lý bệnh-Miễn dịch,
Trường Đại học Y Hà Nội,
Hà Nội, Việt Nam
|
|
FRANCAIS |
|
TURKISH |
Let us know what you think
FEEDBACK |
|
SEARCH |
| |
|
|
 |
|
Logo image © Jeffrey
Nelson, Rush University, Chicago, Illinois and
The MicrobeLibrary |
|
|
|
MỤC TIÊU GIẢNG DẠY
Biết được các thiếu hụt miễn dịch nguyên phát và thứ phát
Biết được tình trạng suy giảm miễn dịch trong bệnh AIDS và
các bệnh khác |
SUY GIẢM MIỄN DỊCH
Suy giảm miễn dịch là sự
thất bại của hệ thống miễn dịch để bảo vệ khỏi bệnh tật hoặc bệnh ác
tính.
Suy giảm miễn dịch
nguyên phát là do các khiếm khuyết di truyền hoặc phát triển trong hệ
thống miễn dịch. Những dị tật này có khi biểu hiện sinh ra nhưng có thể
khi lớn lên mới xuất hiện.
Suy giảm
miễn dịch thứ phát hoặc mắc phải là sự mất chức năng miễn dịch do tiếp
xúc với các tác nhân gây bệnh, các yếu tố môi trường, ức chế miễn dịch
hoặc lão hóa. |
|
Biết được các thiếu hụt miễn dịch nguyên phát chủ yếu và các
đặc điểm của chúng
Hiểu được mối quan hệ giữa vị trí tổn thương và suy giảm miễn
dịch
Biết được các xét nghiệm chẩn đoán các tình trạng suy giảm
miễn dịch khác nhau |
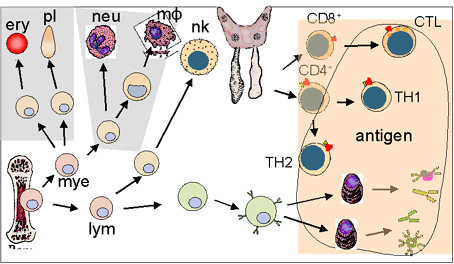
SUY GIẢM MIỄN DỊCH NGUYÊN
PHÁT
Suy giảm miễn dịch nguyên
phát là những khiếm khuyết di truyền của hệ thống miễn dịch (Hình 1). Những
khiếm khuyết này có thể nằm trong các cơ chế miễn dịch đặc hiệu hoặc không
đặc hiệu. Chúng được phân loại dựa trên vị trí tổn thương trong quá trình
phát triển hoặc biệt hóa của hệ thống miễn dịch.
Những người bị suy giảm
miễn dịch dễ bị nhiều loại nhiễm trùng khác nhau và loại nhiễm trùng phụ
thuộc vào bản chất của tình trạng suy giảm miễn dịch (Bảng 1).
|
Bảng 1. Nhiễm trùng đặc trưng của các bệnh thiếu hụt miễn dịch
nguyên phát |
|
Thành phần |
Tác nhân nguyên phát |
Vị trí nguyên phát |
Ví dụ lâm sàng |
|
Tế bào T |
Nội bào, vi khuẩn, virút, đơn bào, nấm |
Không đặc hiệu |
SCID, DiGeorge |
|
Tế bào B |
Pneumococcus,
Streptococcus, Haemophilus
|
Phổi, da, thần kinh trung ương
|
Giảm IgG, IgM
|
|
Vi
khuẩn và virut đường ruột |
Đường
tiêu
hóa, mũi, mắt |
Giảm IgA |
|
Thực bào |
Staphylococcus,
Klebsiella Pseudomonas |
Phổi, da, hạch khu vực |
Bệnh u hạt mạn tính (CGD) |
|
Bổ thể |
Neisseria, Haemophilus,
Pneumococcus, Streptococcus |
Thần kinh trung ương, phổi, da
|
Các thành phần bổ thể C3, Yếu tố I và H, C muộn |
|
|
|
 Hình
1
Hình
1
Các khiếm khuyết xuất hiện trong các bệnh thiếu hụt miễn dịch nguyên phát |
| |
HỆ THỐNG
MIỄN DỊCH ĐẶC HIỆU
Có nhiều
loại suy giảm miễn dịch do khiếm khuyết trong quá trình biệt hóa tế bào
gốc và có thể liên quan đến tế bào T, tế bào B và / hoặc các globulin
miễn dịch thuộc các lớp và phân lớp khác nhau (Bảng 2).
Một khiếm
khuyết trong quá trình tạo máu ban đầu liên quan đến tế bào gốc làm loạn
sinh lưới dẫn đến các khuyết tật miễn dịch nói chung và dễ bị nhiễm
trùng sau đó. Tình trạng này thường gây tử vong nhưng rất hiếm. Nó có
thể được điều trị thành công bằng cách cấy ghép tủy xương.
Suy giảm miễn dịch dòng
lympho
Nếu tế bào tiền thân
của lympho bị lỗi, thì cả dòng tế bào T và B đều bị ảnh hưởng và dẫn
đến tình trạng suy giảm miễn dịch kết hợp nghiêm trọng (SCID). Trẻ
sơ sinh bị các bệnh nhiễm trùng tái phát, đặc biệt là do các vi sinh
vật cơ hội (nhiễm trùng do vi khuẩn, virut, nấm và đơn bào).
Có
khoảng 50% bệnh nhân SCID, tình trạng suy giảm miễn dịch có liên kết
nhiễm sắc thể X trong khi nửa còn lại thì tình trạng suy giảm miễn
dịch là bệnh của nhiễm sắc thể thường. Cả hai đều được đặc trưng bởi
sự vắng mặt của tế bào T và tế bào B và không có (hoặc số lượng rất
thấp) tế bào lympho T và B lưu hành. Bóng tuyến ức không có trên X-quang.
SCID nặng liên kết X
là do khiếm khuyết trong chuỗi gamma của IL-2 và cũng được chia sẻ
bởi IL-4, -7, -11 và 15, tất cả đều liên quan đến sự tăng sinh và /
hoặc biệt hóa tế bào lympho. Các SCID nhiễm sắc thể thường phát sinh
chủ yếu từ các khiếm khuyết trong gen adenosine deaminase (ADA) hoặc
purine nucleoside phosphorylase (PNP), kết quả là sự tích tụ của
dATP hoặc dGTP tương ứng và gây ra độc tính đối với tế bào gốc
lympho.
Các khiếm khuyết di
truyền khác dẫn đến SCID bao gồm những khiếm khuyết của RAG1, RAG2
và IL-7-alpha. Nếu nghi ngờ SCID, bệnh nhân không được tiêm vaccin
sống, vì nó sẽ dẫn đến bệnh tiến triển.
Chẩn
đoán dựa trên việc đếm các tế bào T và B và đo lượng globulin miễn
dịch. Suy giảm miễn dịch kết hợp nghiêm trọng có thể được điều trị
bằng cách cấy ghép tủy xương (xem MHC và cấy ghép). Gần đây, có một
số thành công khi các bệnh nhân SCID thể nhiễm sắc thể thường thiếu
ADA được điều trị bằng vectơ retrovirus chuyển gen.
SCID gồm
một
số
rối
loạn
nặng
Gen hoạt hóa tái tổ hợp
Bệnh nhân bị thiếu hụt cả tế bào T và B đều thiếu các gen hoạt hóa
tái tổ hợp (RAG1 và 2) chịu trách nhiệm tái sắp xếp thụ thể tế bào T
và gen globulin miễn dịch. Những bệnh nhân này có sức khỏe dẻo dai
và được chẩn đoán bằng cách kiểm tra sự sắp xếp lại gen của thụ thể
tế bào T (TCR). Các khiếm khuyết trong tế bào B không được quan sát
thấy trong giai đoạn đầu đời của trẻ sơ sinh vì các kháng thể thụ
động thu được từ mẹ. Tế bào NK bình thường ở những bệnh nhân này.
Đây là một tính trạng lặn trên NST thường.
Chuỗi CD3
Ở một số bệnh nhân SCID, tế bào T có thể tồn tại
nhưng khiếm khuyết về mặt chức năng do thiếu hụt tín hiệu qua trung
gian chuỗi CD3 có liên quan đến TCR.
Thụ thể interleukin-2
Chuỗi gamma chung của thụ thể interleukin-2 (IL-2Rγc)
có thể bị thiếu ở bệnh nhân, do đó ngăn cản tín hiệu của IL-2 và các
cytokin khác hoạt động như các yếu tố tăng trưởng. Điều này dẫn đến
khiếm khuyết trong quá trình tăng sinh của tế bào T, tế bào B và tế
bào NK. Đây là một tính trạng lặn trên NST thường.
Adenosine deaminase
Adenosine deaminase (ADA) là một loại enzym chịu
trách nhiệm chuyển đổi adenosin thành inosin. Sự thiếu hụt ADA dẫn
đến tích tụ adenosin, dẫn đến việc sản xuất các chất chuyển hóa độc
hại cản trở quá trình tổng hợp DNA. Bệnh nhân có khiếm khuyết trong
tế bào T, B và NK.
SCID
là các đặc điểm lặn trên NST thường và có thể được điều trị bằng
liệu pháp gen hoặc cấy ghép tế bào gốc.
|
Bảng 2. Tóm tắt các bệnh suy giảm miễn dịch tế bào T và tế
bào B (ID) |
|
Bệnh |
Tế bào T |
Tế bào B
No |
Kháng thể |
Di truyền |
|
No. |
Fx |
IgM |
IgG |
IgA |
|
Rối loạn chức năng lưới |
A |
A |
A |
A |
A |
A |
u |
|
CID (NST thường) |
A/L |
A/L |
A/L |
A/L |
A/L |
A/L |
a |
|
SCID (Liên kết x) |
A/L |
A/L |
A/L |
A/L |
A/L |
A/L |
x |
|
Hội chứng DiGeorge |
A/L |
A/L |
N/V |
N/V |
N/V |
N/V |
a/x |
|
Thất điều giãn mạch |
L |
L |
L |
N/V |
L/V |
L |
a
|
|
Wiskott-Aldrich
|
?V |
L |
L/V |
L |
N |
H |
x |
|
IgE cũng cao |
|
Giảm gamma- globulin máu kiên kết X |
N |
N |
L |
L |
L |
L |
x |
|
Giảm IgA chọn lọc |
N |
N |
N |
N |
L/V |
L |
a/x |
|
Giảm gamma- globulin máu tăng IgM |
N |
N |
N |
H |
L |
L |
x |
|
Giảm gamma-globulin máu thoáng qua |
N |
N |
N |
N |
L |
L |
a? |
|
Giảm gamma-globulin máu thay đổi phổ biến (thanh thiếu
niên-người lớn) |
N |
N |
N |
N |
L |
L |
không |
|
A: vắng mặt; a: NST thường; H: cao; L: thấp; N: bình thường;
U; không xác định; V: biến; x: liên kết x |
|
|
|
Rối loạn tế bào T
Rối loạn tế bào T ảnh
hưởng đến cả miễn dịch qua trung gian tế bào và dịch thể, làm cho bệnh
nhân dễ bị nhiễm virut, đơn bào và nấm. Những bệnh nhiễm trùng do virut
như cytomegalovirus và sởi giảm độc lực trong vaccin có thể gây tử vong
ở những bệnh nhân này.
Hội
chứng DiGeorge (Hội chứng mất NST 22)
Đây là tình trạng suy giảm miễn dịch tế bào T được
xác định rõ ràng nhất và còn được gọi là bất sản / giảm sản tuyến ức
bẩm sinh, hoặc suy giảm miễn dịch kèm theo suy tuyến cận giáp. Hội
chứng có liên quan đến suy tuyến cận giáp, bệnh tim bẩm sinh, tai có
khía thấp và miệng hình cá. Những dị tật này là kết quả của sự phát
triển bất thường của thai nhi (túi hầu họng thứ 3 và thứ 4) trong
tuần thứ 6 đến tuần thứ 10 của thai kỳ khi tuyến cận giáp, tuyến ức,
môi, tai và cung động mạch chủ đang được hình thành. Không có khuynh
hướng di truyền nào rõ ràng và không phải tất cả trẻ sơ sinh mắc hội
chứng DiGeorge đều bị bất sản tuyến ức. Có thể sử dụng mảnh ghép
tuyến ức từ thai nhi sớm (13 - 14 tuần tuổi) để điều trị. Các mảnh
ghép muộn hơn có thể dẫn đến phản ứng GVH. Ở những bệnh nhân
DiGeorge bị suy giảm miễn dịch nghiêm trọng, vaccin sống có thể gây
nhiễm trùng tiến triển.
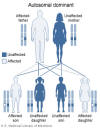
Hội chứng DiGeorge
là bệnh trội NST thường (Hình 2) và do mất đoạn nhiễm sắc thể 22 (Hình
3). Sự mất đoạn có thể có kích thước khác nhau nhưng nó không tương
quan với mức độ nghiêm trọng của bệnh. Trong khoảng 6% trường hợp,
mất vi đoạn nhiễm sắc thể 22 được di truyền nhưng hầu hết các trường
hợp là do mất đoạn de novo có thể do các yếu tố môi trường
gây ra. Bệnh nhân có thể được điều trị bằng phương pháp ghép tuyến
ức.
|
|
CASE
PRESENTATION
Pediatric
Pathology
DiGeorge Syndrome
A 24-day-old Term Infant with Seizures
(Department of Pathology, University of Pittsburgh) |
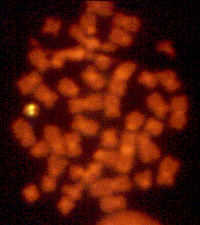
 Hình 2
Hình 2
Trong hội chứng DiGeorge, sự mất đoạn 22q11.2 được di truyền theo kiểu trội
trên NST thường.
Thư viện Y khoa Quốc gia – NIH
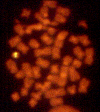 Hình 3
Hình 3
Việc mất các gen trong hội chứng DiGeorge có thể được phát hiện bằng tín
hiệu huỳnh quang chỉ trên một trong hai bản sao của nhiễm sắc thể 22.
David
Ian Wilson, Đại học Newcastle trên Tyne - NIH |
Suy giảm tế bào T với mức
độ suy giảm tế bào B khác nhau
Hội
chứng thất điều giãn mạch
Hội chứng thất
điều giãn mạch là sự thiếu hụt các tế bào T liên quan đến sự thiếu
phối hợp vận động (mất điều hòa) và giãn nở các mạch máu nhỏ của
vùng mặt (telangiectasis). Tế bào T và chức năng của chúng bị giảm ở
nhiều mức độ khác nhau. Số lượng tế bào B và nồng độ IgM từ bình
thường đến thấp. IgG thường giảm và IgA giảm đáng kể (trong 70%
trường hợp). Ở những bệnh nhân này bệnh ách tính có thể xuất hiện ở
một tỷ lệ cao, đặc biệt là bệnh lơ xê mi. Các khiếm khuyết phát sinh
do sự đứt gãy ở nhiễm sắc thể 14 tại vị trí của các gen TCR và gen
chuỗi nặng globulin miễn dịch.
Hội
chứng Wiskott-Aldrich
Hội chứng Wiskott-Aldrich
có liên quan đến số lượng tế bào T bình thường nhưng các chức năng
bị suy giảm, tiến triển nặng hơn. Nồng độ IgM giảm nhưng nồng độ IgG
vẫn bình thường. Cả hai nồng độ IgA và IgE đều tăng cao. Các bé trai
mắc hội chứng này phát triển thành bệnh chàm nặng, chấm xuất huyết
(do khiếm khuyết tiểu cầu và giảm tiểu cầu). Chúng phản ứng kém với
các kháng nguyên polysaccharid và dễ bị nhiễm trùng sinh mủ. Hội
chứng Wiskott-Aldrich là một rối loạn liên kết X (Hình 4) do khiếm
khuyết glycoprotein của khung tế bào, CD43.
Suy
giảm MHC (Hội chứng bạch cầu trần)
Một số trường hợp
suy giảm miễn dịch đã được mô tả trong đó có khiếm khuyết trong gen
protein điều hòa phiên mã MHC lớp II (CIITA), dẫn đến thiếu phân tử
MHC lớp II trên APC của chúng. Vì sự chọn lọc dương của các tế bào
CD4 trong tuyến ức phụ thuộc vào sự hiện diện của các phân tử MHC
này, những bệnh nhân này có ít tế bào CD4 hơn và dễ bị nhiễm trùng.
Cũng có những cá nhân bị khiếm khuyết trong gen liên kết vận chuyển
protein (TAP) và do đó không biểu hiện các phân tử MHC lớp I và do
đó thiếu tế bào T CD8 +.
|
 Hình 4
Hình 4
Hội chứng Wiskott-Aldrich là một chứng rối loạn liên kết X
Thư viện Y
khoa Quốc gia - NIH |
Rối loạn tế bào lympho B
Có một số bệnh mà số lượng và chức năng của tế bào T vẫn
bình thường: Số lượng tế bào B có thể thấp hoặc bình thường nhưng lượng
globulin miễn dịch thấp.
Giảm
globulin miễn dịch ở trẻ sơ sinh liên kết X
Giảm globulin miễn
dịch liên kết X, còn được gọi là bệnh Bruton hoặc giảm
globulin huyết, là tình trạng giảm globulin miễn dịch huyết
nghiêm trọng nhất, trong đó số lượng tế bào B và tất cả các nồng độ
globulin miễn dịch đều rất thấp. Các bệnh nhân bị suy giảm quá trình
trưởng thành của tế bào B liên quan đến gen tyrosine kinase (btk)
của tế bào B bị lỗi. Do đó, tế bào B tồn tại dưới dạng tiền tế bào B
với chuỗi H nhưng không phải chuỗi L được sắp xếp lại. Chẩn đoán dựa
trên việc đếm tế bào B và đo lượng globulin miễn dịch. Bệnh nhân
không có globulin miễn dịch và dễ bị nhiễm trùng tái phát do vi
khuẩn.
Giảm
globulin miễn dịch thoáng qua
Trẻ em khi mới sinh có nồng độ IgG tương đương với
nồng độ IgG của người mẹ. Vì thời gian bán hủy của IgG là khoảng 30
ngày, mức độ của nó giảm dần, nhưng đến ba tháng tuổi, trẻ sơ sinh
bình thường bắt đầu tổng hợp IgG của riêng mình. Tuy nhiên, ở một số
trẻ sơ sinh, quá trình tổng hợp IgG có thể không bắt đầu cho đến khi
chúng được 2 đến 3 tuổi. Sự chậm trễ này được cho là do sự hỗ trợ
của tế bào T kém. Điều này dẫn đến sự thiếu hụt tạm thời IgG có thể
được điều trị bằng gamma-globulin.
Giảm
globulin miễn dịch biến đổi thường gặp (Giảm globulin miễn dịch khởi
phát muộn)
Những người này bị
thiếu hụt IgG và IgA trong thập kỷ thứ 2 hoặc thứ 3 của cuộc đời vì
tế bào B không thể biệt hóa thành tương bào. Những bệnh nhân này dễ
bị nhiễm nhiều loại vi khuẩn sinh mủ và đơn bào đường ruột. Chúng
nên được điều trị bằng gamma-globulin được điều chế đặc biệt để sử
dụng cho đường tĩnh mạch.
Thiếu hụt IgA
Thiếu IgA là tình
trạng phổ biến nhất của tất cả các trường hợp thiếu hụt miễn dịch
(1/700 trong số tất cả người da trắng) và là kết quả của việc chuyển
đổi lớp kháng thể. Khoảng 20%
người
thiếu IgA cũng có IgG thấp. Bệnh nhân thiếu IgA rất dễ bị nhiễm
trùng đường tiêu hóa, mắt và mũi họng. Bệnh nhân thiếu hụt IgA có tỷ
lệ mắc các bệnh tự miễn dịch cao (đặc biệt là loại phức hợp miễn
dịch) và các khối u ác tính lympho. Kháng thể kháng IgA (IgG) được
phát hiện ở 30% đến 40% bệnh nhân không nên điều trị bằng
γ-globulin. Chẩn đoán trong phòng thí nghiệm dựa trên xét nghiệm IgA.
Thiếu hụt IgG có chọn lọc
Người ta đã tìm thấy sự suy giảm của các phân lớp IgG
khác nhau. Những bệnh nhân này dễ bị nhiễm trùng sinh mủ.
Suy
giảm miễn dịch liên kết X có tăng IgM
Những người mắc
loại suy giảm miễn dịch này có nồng độ IgA và IgG thấp; IgM cao bất
thường. Những bệnh nhân này không thể chuyển từ IgM sang các lớp
khác được là do khiếm khuyết CD40L trên tế bào CD4 của họ. Chúng rất
dễ bị nhiễm trùng sinh mủ và cần được điều trị bằng gamma-globulin
tiêm tĩnh mạch.
|
 Hình 5
Hình 5
Tiêu diệt vi khuẩn nội bào kém trong bệnh u hạt mãn tính
|
HỆ THỐNG MIỄN DỊCH KHÔNG
ĐẶC HIỆU – SUY GIẢM DÒNG TỦY
Suy giảm miễn dịch nguyên phát của hệ thống miễn dịch
không đặc hiệu bao gồm những khiếm khuyết trong tế bào thực bào, NK và
hệ thống bổ thể.
Giảm bạch cầu hạt trung tính bẩm
sinh
Bệnh nhân bị giảm
số lượng bạch cầu trung tính. Điều này là do khiếm khuyết trong quá
trình biệt hóa tế bào tiền thân dòng tủy thành bạch cầu trung tính.
Những bệnh nhân này được điều trị bằng yếu tố kích thích dòng bạch
cầu hạt-đại thực bào (GM-CSF) hoặc G-CSF.
Khiếm khuyết của hệ thống thực bào
Sự khiếm khuyết của các tế bào thực bào (số lượng và
/ hoặc chức năng) có thể dẫn đến tăng tính nhạy cảm với nhiều loại
bệnh nhiễm trùng.
Giảm bạch cầu trung tính theo chu kỳ
Điều này được thể
hiện là số lượng bạch cầu trung tính lưu hành thấp khoảng ba tuần
một lần. Tình trạng giảm bạch cầu kéo dài khoảng một tuần, trong đó
bệnh nhân dễ bị nhiễm trùng. Sự khiếm khuyết dường như là do sự điều
hòa kém của việc sản xuất bạch cầu trung tính.
Bệnh u hạt mãn tính (CGD)
CGD
được đặc trưng bởi hạch to rõ rệt, gan - lách to và các hạch bạch
huyết thoát dịch mãn tính. Bạch cầu có khả năng tiêu diệt nội bào
kém (Hình 5) và sự bùng phát hô hấp tế bào thấp. Phần lớn sự thiếu
hụt ở những bệnh nhân này là do khiếm khuyết NADPH oxidase (cytochrome
b558: gp91phox, hoặc hiếm khi là gp22phox) hoặc các protein phối hợp
khác (gp47phox, gp67phox) tham gia vào quá trình bùng nổ hô hấp thực
bào. Những bệnh nhân này có thể được chẩn đoán trên cơ sở giảm
Nitroblue tetrazolium (NBT) kém, đây là một biện pháp của sự bùng nổ
hô hấp. Liệu pháp interferon-gamma đã thành công.
|
 Hình 6
Hình 6
Hình này là của một bệnh nhân mắc Hội chứng Chediak-Higashi. Các hạt cực lớn
được nhìn thấy trong tế bào chất của bạch cầu hạt. Chúng là kết quả của sự
hợp nhất bất thường của các hạt trong quá trình hình thành của chúng. Các
hạt bất thường được tìm thấy trong nhiều loại tế bào khác trên khắp cơ thể.
Viện ung
thư quốc gia |
Sự suy giảm kết dính bạch cầu
Trong bệnh này, tế bào
T và đại thực bào thiếu thụ thể bổ thể CR3 do khiếm khuyết trong peptid
CD11 hoặc CD18 và do đó chúng không thể đáp ứng với opsonin C3b. Ngoài
ra, có thể có khiếm khuyết trong các phân tử kết dính, LFA-1 hoặc mac-1
phát sinh từ các peptid CD11a hoặc CD11b bị lỗi, tương ứng. Các phân tử
này tham gia vào quá trình xuyên mạch và do đó các bạch cầu trung tính
bị lỗi không thể phản ứng hiệu quả với các tín hiệu hóa học. Điều trị
bằng cách cấy ghép tủy xương (không có tế bào T và phù hợp với MHC) hoặc
liệu pháp gen.
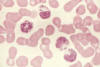
Hội chứng Chediak-Higashi
Hội chứng Chediak-Higashi
được đánh dấu bằng việc giảm (tốc độ chậm hơn) sự tiêu diệt nội bào và
hóa hướng động kèm theo không có khả năng dung hợp phagosom và lysosom
và suy giảm proteinase. Các lysosom khổng lồ (các hạt nội bào) thường
được nhìn thấy (Hình 6). Sự bùng nổ hô hấp diễn ra bình thường. Các
khuyết tật tế bào NK kèm theo và rối loạn tiểu cầu và thần kinh được ghi
nhận.
|
| |
RỐI LOẠN HỆ THỐNG BỔ THỂ
Các bất thường về bổ thể cũng
dẫn đến tăng nhạy cảm với các bệnh nhiễm trùng. Có sự thiếu hụt di truyền
của các thành phần khác nhau của hệ thống bổ thể, trong đó nghiêm trọng nhất
là sự thiếu hụt C3 có thể phát sinh do tổng hợp C3 thấp hoặc thiếu hụt yếu
tố I hoặc yếu tố H.
|
| |
SUY GIẢM MIỄN DỊCH THỨ
PHÁT (MẮC PHẢI)
Suy giảm miễn dịch
liên quan đến nhiễm trùng
Nhiễm trùng do vi
khuẩn, virut, đơn bào, sán và nấm có thể dẫn đến suy giảm tế bào B, tế
bào T, PMN và đại thực bào. Nổi bật nhất trong số này là hội chứng suy
giảm miễn dịch mắc phải (AIDS). Suy giảm miễn dịch thứ phát cũng được
phát hiện trong các bệnh ung thư.
Bất thường miễn dịch
trong AIDS
Tất cả các loại suy
giảm miễn dịch mắc phải đã bị qua mặt bởi AIDS do virut gây suy giảm
miễn dịch ở người (HIV) -1. Loại virus này được phát hiện lần đầu tiên
vào năm 1981 và các bệnh nhân có biểu hiện nhiễm nấm với các sinh vật cơ
hội như Pneumocystis carinii và trong các trường hợp khác, với một khối
u da được gọi là sarcoma Kaposi. Có hai loại HIV chính: HIV-1 và 2, loại
trước đây là chủng thường được tìm thấy ở Bắc Mỹ. HIV lây lan qua quan
hệ tình dục, máu và dịch cơ thể bị nhiễm bệnh cũng như từ mẹ sang con.
HIV là một loại virus retrovirus có RNA được phiên mã ngược thành DNA
bằng enzym transciptase (RT) sau khi xâm nhập vào tế bào. DNA được tích
hợp vào bộ gen tế bào như một provirus được sao chép cùng với tế bào.
HIV-1 không tái tạo ở hầu hết các loài động vật khác nhưng lây nhiễm
sang tinh tinh mặc dù nó không gây ra bệnh AIDS ở chúng. Những con chuột
bị suy giảm miễn dịch phối hợp nặng (SCID) được hoàn nguyên bằng tế bào
lympho của người có thể bị nhiễm HIV-1. Hạt virut HIV-1 bao gồm một vỏ
virut được tạo thành từ lớp kép lipid bên ngoài của tế bào chủ, trong đó
có các glycoprotein nhúng bao gồm gp41 xuyên màng cùng với gp120 liên
kết. Gp120 liên kết CD4 được thể hiện trên các tế bào chủ. Bên trong vỏ
virut là lõi virut hoặc nucleocapsid bao gồm một lớp protein nền bao gồm
p17 và capsid bên trong được tạo thành từ p24. Bộ gen của virut bao gồm
hai phân tử RNA sợi đơn liên kết với hai phân tử enzym sao chép ngược (RT)
cũng như các enzym khác bao gồm một protease và một integrationse.
Chu
kỳ nhân lên và đích của điều trị
Virut gắn vào phân
tử CD4 trên tế bào Th, bạch cầu đơn nhân và tế bào có tua thông qua
gp120 của HIV. Đối với nhiễm HIV, cần phải có đồng thụ thể. Đồng thụ
thể là một thụ thể chemokin như CXCR4 hoặc CCR5. CCR5, biểu hiện chủ
yếu trên đại thực bào, và CXCR4 trên tế bào T CD4 đóng vai trò là
chất thụ cảm cốt lõi để lây nhiễm HIV. Sau khi hợp nhất vỏ HIV và
màng vật chủ, nucleocapsid đi vào tế bào. RT tổng hợp DNA của virut
được vận chuyển đến nhân nơi nó kết hợp với DNA của tế bào dưới dạng
một provirus. Provirus có thể tồn tại tiềm ẩn cho đến khi tế bào
được kích hoạt khi provirus cũng trải qua quá trình phiên mã. Hạt
virut, bao gồm RNA và protein của virut đã phiên mã, được tạo ra.
Những chồi này ra khỏi màng tế bào chủ, nơi chúng thoát vỏ. Do đó,
các tác nhân điều trị đã được phát triển nhằm mục tiêu sự xâm nhập
và dung hợp của virut, cũng như đóng vai trò như các chất ức chế RT,
protease và tích hợp gen. Liệu pháp kháng hoạt động của virut cao là
một hỗn hợp của 3 hoặc nhiều chất như vậy.
Thay
đổi miễn dịch học
Virut nhân lên nhanh chóng và trong vòng khoảng hai
tuần, bệnh nhân có thể bị sốt. Tải lượng virut trong máu tăng đáng
kể và đạt đỉnh điểm trong hai tháng, sau đó giảm đột ngột vì virut
tiềm ẩn được tìm thấy trong các trung tâm mầm của các hạch bạch
huyết. CTL phát triển rất sớm trong khi các kháng thể có thể được
phát hiện từ 3 đến 8 tuần. CTL giết chết tế bào Th trong khoảng 4-8
tuần dẫn đến giảm tế bào T CD4. Khi số lượng tế bào T CD4 giảm xuống
dưới 200 mỗi mm khối, bệnh AIDS phát triển hoàn toàn.
Liệu pháp miễn dịch
Có một số khó khăn để phát triển vaccin HIV hiệu
quả.
-
Vaccin giảm
độc lực có thể gây ra bệnh
-
Các tế bào T
CD4 có thể bị phá hủy bởi vaccin
-
Sự biến đổi
kháng nguyên của HIV
-
Khả năng sinh
miễn dịch của virut thấp bằng cách giảm biểu lộ các phân tử
MHC
-
Thiếu mô hình
động vật
-
Thiếu các xét
nghiệm in vitro
Các chất sau đây đã được xem xét trong việc phát
triển vaccin
-
Mẫn cảm với
các đột biến mất đoạn để giảm khả năng gây bệnh
-
Tiêm chủng
bằng các protein tái tổ hợp
-
Các protein
mã hóa của gen được đưa vào vectơ virut có thể được sử dụng
để tiêm chủng
-
Các chemokin
cạnh tranh cho các đồng thụ thể
-
IL-2 để tăng
cường các tế bào Th.
Để biết thêm
về HIV và AIDS, hãy vào
đây
Suy giảm miễn dịch
liên quan đến lão hóa
Chúng bao gồm giảm
dần vỏ tuyến ức, giảm mô tế bào và giảm kích thước của tuyến ức,
giảm chức năng tế bào ức chế và do đó tăng tự phản ứng, giảm chức
năng của tế bào CD4. Ngược lại, các chức năng của tế bào B có thể
được nâng lên một chút.
Suy giảm miễn
dịch liên quan đến khối u ác tính và các bệnh khác
Thiếu hụt tế bào B
đã được ghi nhận trong bệnh đa u tủy, bệnh macroglobulin máu
Waldenstrom, bệnh lơ xê mi dòng lympho mạn tính và u lympho biệt hóa
tốt. Bệnh Hodgkin và các khối u rắn tiến triển có liên quan đến chức
năng tế bào T bị suy giảm. Hầu hết các chất hóa trị liệu được sử
dụng để điều trị các khối u ác tính cũng có tác dụng ức chế miễn
dịch.
Các điều kiện khác
mà tình trạng suy giảm miễn dịch thứ phát xảy ra là thiếu máu hồng
cầu hình liềm, đái tháo đường, suy dinh dưỡng calo protein, bỏng, xơ
gan do rượu, viêm khớp dạng thấp, suy thận, v.v.
|
|
|
 Return to the Immunology Section of Microbiology and Immunology On-line
Return to the Immunology Section of Microbiology and Immunology On-line
This page last changed on
Wednesday, September 02, 2020
Page maintained by
Richard Hunt
|
 Hình
1
Hình
1 Hình 2
Hình 2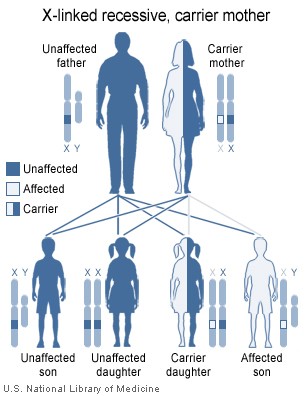 Hình 4
Hình 4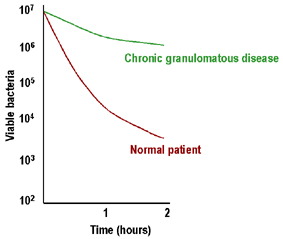 Hình 5
Hình 5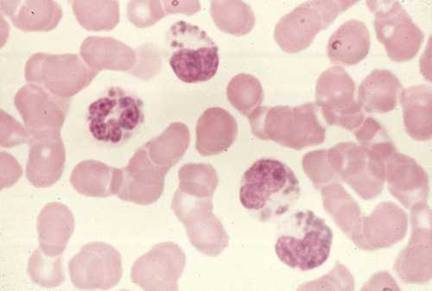 Hình 6
Hình 6