![]()

VIROLOGY - CHAPTER TWENTY THREESLOW VIRUS DISEASES OF THE NERVOUS SYSTEM
Dr Margaret Hunt
Professor Emerita
Department of Pathology, Microbiology and Immunology
University of South Carolina School of Medicine
Columbia
FEEDBACK
Edited and illustrated by Dr Richard Hunt
TEACHING OBJECTIVES
Introduction to subacute/chronic diseases of the central nervous system.
Properties of unconventional agents associated with subacute diseases of the central nervous system
GENERAL
The term "Slow virus infections" refers to the tempo of the DISEASE, not to the growth rate of the virus. These diseases have a prolonged incubation period (which can be months or years), and a protracted, progressive clinical course.
Slow virus diseases may be caused by conventional viruses or by the unconventional viruses (also known as the unconventional agents or atypical viruses/agents).
The symptoms associated with slow viral/prion diseases of the central nervous system tend to have multiple neurological manifestations. Different patients may present with very different symptoms.
Figure 1
Brain in progressive multifocal leucoencephalopathy
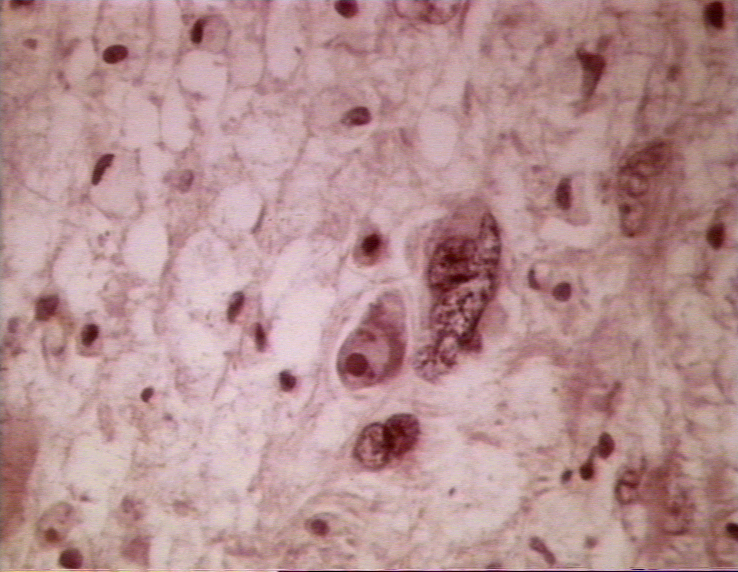 Figure 2
Figure 2
Brain in progressive multifocal leucoencephalopathy ©
Bristol Biomedical Image Archive. Used with permission
CONVENTIONAL VIRUSES
Progressive multifocal leukoencephalopathy (PML)
This is a rare, progressive, fatal, demyelinating disease of the CNS that kills oligodendrocytes (figure 1 and 2). It results in memory loss, loss of co-ordination, mentation problems, vision problems, etc.
The disease is caused by certain members of the polyomavirus family, usually JC virus. Serology shows that exposure to JC virus is common, but PML is rare. Patients who develop PML frequently have some abnormality of the immune system. PML develops in up to 5% of patients with AIDS. In AIDS, HAART treatment may be able to stabilize at least some of these patients and their neuroradiological picture may even improve. However, not all HIV-positive PML patients show an obvious response to HAART with respect to PML. PML may be due to reactivation of a JC virus latent infection, probably in the kidney. There is also abundant virus in brain.
Another polyoma virus, BK virus, can establish a latent infection in the kidney and under conditions of immune suppression be reactivated and may cause severe urinary tract infections. It is not associated with PML. There has been a suggestion (2006) that BK virus might play a role in prostate cancer.
Subacute sclerosing panencephalitis (SSPE)
This disease is a rare complication of measles virus infection and develops approximately 1 to 10 years after the initial infection. It is progressive and fatal and is characterized by mental and motor deterioration. Risk factors include acquiring primary measles at an early age.
SSPE is associated with defective forms of the virus in the brain and so it is difficult to isolate infectious virus from patients. Incidence has decreased since the introduction of anti-measles vaccination.
Progressive rubella panencephalitis (PRP)
PRP is a very rare consequence of rubella virus infection and also results in mental and motor deterioration. The initial infection is usually congenital or soon after birth and the onset of PRP occurs at 8 to 19 years of age. The course of the disease may extend over many years.
Other slow virus infections
Human immunodeficiency virus and AIDS. See HIV/AIDS section
Rabies. See rabies section
PRIONS
 Figure
3
Figure
3Comparison of the properties of conventional viruses and prions
Some slow diseases of the central nervous system are caused by a group of unusual agents, whose true nature is still controversial. In some ways the agents are like conventional viruses: They are very small, filterable agents that require host cells to grow. They have no capacity for energy generation or protein synthesis.
In other ways, however, they are rather different from viruses; for example, we cannot see any evidence of virus particles in infected tissues or purified preparations of infectious material. No one has been able to prove that these agents contain nucleic acid. If they contain nucleic acid, it is likely that it is very small and has very little coding capacity. These agents have an unusual resistance to treatments commonly used to inactivate viruses (figure 3 and 4).
 Figure
4
Figure
4
Properties of unconventional viruses
 Figure 5
Figure 5
Transmissible spongiform encephalopathies
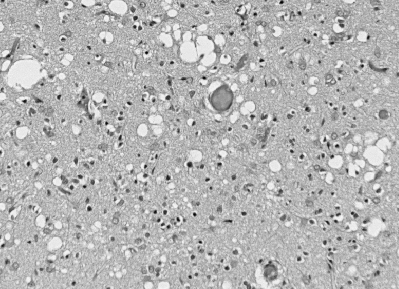 Figure 6
Figure 6
Spongiform change in CJD consists of numerous rounded vacuoles within the neuropil which occur both singly and in confluent groups, distorting the cortical
cytoarchitecture. © The UK Creutzfeldt-Jakob Disease Surveillance Unit
These unconventional viruses or agents are often called 'prions' - since protein is present in purified preparations of infectious material and treatments that destroy protein destroy infectivity. In contrast, treatments that destroy nucleic acids do not destroy infectivity. This protein is known as PrP (prion protein). The question as to whether nucleic acids are part of these agents is still controversial; many people think that the infectious material is protein only.
These agents:
cause diseases that are confined to the CNS
have a prolonged incubation period
show a slow, progressive, fatal course of disease
show a spongiform encephalopathy
characteristically result in vacuolation of neurons
can cause formation of fibrillar aggregates, which contain PrP and have amyloid-like characteristics
Diseases caused by these agents (transmissible spongiform encephalopathies (figure 5)) are relatively rare in man, but there is speculation that they may be more common than previously thought and they may have implications in the study of other CNS degenerative diseases. They may be acquired, inherited, or occur sporadically.
Animal Prion-caused diseases
Scrapie
Scrapie is a disease of sheep. It results in behavioral changes, progresses to tremor, ataxia (failure of muscle coordination), wasting and death. It is a transmissible disease.
Bovine Spongiform Encephalitis (BSE)
BSE, otherwise known as mad cow disease is a prion-caused disease that results in progressive neurological degeneration in cattle. The first cases were identified in 1986 in the United Kingdom with infections probably having occurred in the 1970’s. Transmission probably occurred as a result of feeding cows with meat and bone meal from other BSE-infected cows (an infection that might have arisen spontaneously) or with meat and bone from Scrapie-infected sheep. Control of the disease has resulted from banning this practice.
BSE peaked in the United Kingdom in 1992 when new cases were being reported at more than 1,000 per week but because of control measures, the rate of new infections has fallen to 1 to 2 per year.
In North America, there have been 24 cases of BSE (through February 2015) of which 20 have been in Canada
WEB RESOURCES
NINDS
Kuru Information Page
Creutzfeldt-Jakob Disease Fact Sheet
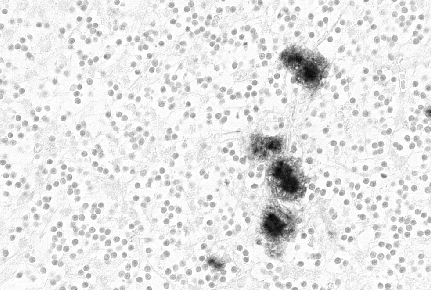 Figure 7
Figure 7Prion protein immunostaining showing amyloid plaques. © The UK Creutzfeldt-Jakob Disease Surveillance Unit
Human Prion-caused diseases
Kuru
Kuru is a disease of man. It causes tremors and ataxia and often, in later stages, dementia. It is transmitted by rites for the dead which included autopsy and cannibalism in Fore people in Papua/New Guinea. No one born since these practices ceased has acquired Kuru. There is no evidence for transmission to fetus, transmission via milk or intimate social contact.
Creutzfeldt-Jakob disease (CJD)
CJD is a disease of man resulting in dementia and also often tremors and lack of motor co-ordination. In the United States, there are 1 to 2 cases per million population per annum but cases may go undiagnosed. The disease can be transmitted to animals in the laboratory. Although the disease have been observed to develop at ages 16 to 80+ years, it is usually seen at 50 to 70 years of age. 10% of cases are familial suggesting that a gene apparently makes the individual more likely to develop CJD.
The usual means of transmission is not known but most cases are sporadic and there is no evidence for direct person-person transmission. CJD can be transmitted by medical manipulations: cornea transplants, dura mater transplants, use of improperly sterilized equipment in neurosurgery (sterilization procedures have now been changed to prevent this), human cadaver growth hormone administration (recombinant DNA vector is now used to make human growth hormone).
WEB RESOURCES
BSE
CDC
; nvCJD, vCJDNew variant CJD disease (human BSE)
A new form of CJD was reported in 1996, predominantly in the United Kingdom, in patients who were usually younger (frequently under 40; average age at death: 28 years) than is the case for most CJD patients (average age of death: 68 years) (figure 8A). This disease was also different from the usual CJD in that patients tended to present with psychiatric problems and in that the course of the disease tended to be more protracted. This disease is known as variant Creutzfeldt-Jakob disease or vCJD.
vCJD patients may eventually show any or all of the symptoms described above for other human prion diseases and there is strong evidence to suggest that it is associated with exposure to BSE-contaminated beef. Strong BSE control measures have now been implemented. Autopsy reveals a distinctive neuropathological appearance and more PrP (prion protein) amyloid plaque type deposits (figure 7 and 9) than in typical CJD cases.
| Differences between variant CJD and classical CJD | Number of BSE cases |
As of 2014, there have been over 220 cases of vCJD reported around the world. 177 people have been reported with vCJD in the UK, all of whom have died (figure 8b), 27 in France, 4 in Ireland, 4 in the United States and 1 each in Canada, Japan, Portugal, Spain and the Netherlands (the people from Canada, Japan and the US were probably exposed while living in the UK). The peak year of the UK epidemic was 2000 when there were 27 people diagnosed with vCJD and 28 deaths from the disease.
We do not know if we are seeing the beginning of a major outbreak or whether these will be the majority of cases of this disease ever seen. Further peaks are possible in different genetic groups but there has, so far, been no evidence of another wave of vCJD. There is also concern because there appears to be more infectious agent in the peripheral tissues, especially the lymphoreticular tissue of patients with vCJD than with normal CJD. This raises questions about sterilization of surgical instruments etc. and the possibility of iatrogenic spread. This is also one of the reasons that the United States is concerned and conservative about protecting the blood supply (by screening out those who have spent considerable time in the UK or Europe). There is a possibility that vCJD agents may have been transmitted by blood transfusion in two cases in the United Kingdom. In both cases, the blood had not been leuko-depleted; it is thought leuko-depletion might decrease the chances of transmission by blood transfusion.
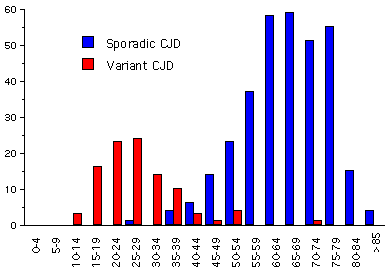 Figure 8A
Figure 8AAge Distribution of CJD in UK: CJD and vCJD 1994-2001 USDA
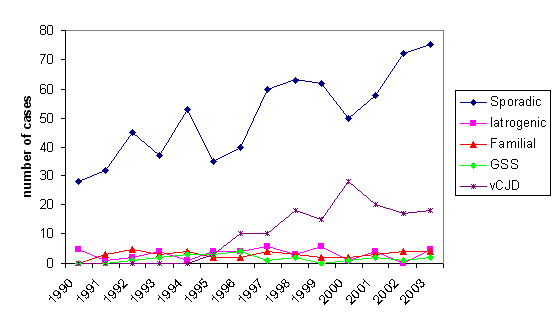 Figure 8B
Figure 8BNumber of deaths due to CJD in UK 1990-2003
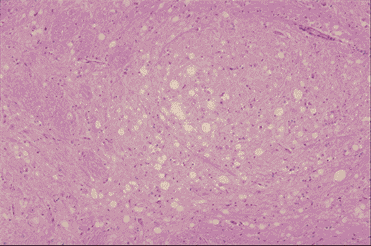
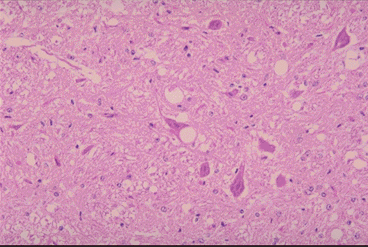 Figure 9A
Figure 9A
Medulla of the BSE affected cow: Vacuoles are seen in one neuron and in the
neuropiles. Astrocytes with small
nucleus proliferate. No inflammatory cells infiltrae in the brain. Left x100, Right x200
Dr. M. KUBO NIAH, Japan
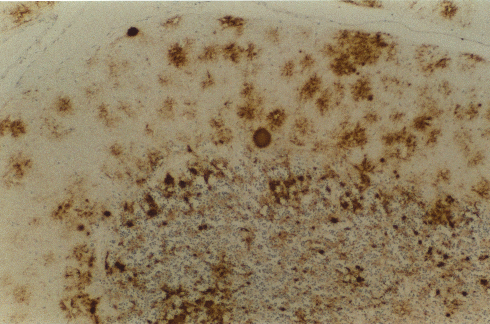 Figure 9B
Figure 9B
Immunocytochemistry for PrP in the cerebellum shows strong staining of a
kuru- type plaque (centre) with multiple smaller plaques in the granular layer and abundant pericellular deposition in the molecular layer
© The UK Creutzfeldt-Jakob Disease Surveillance Unit/The
Lancet
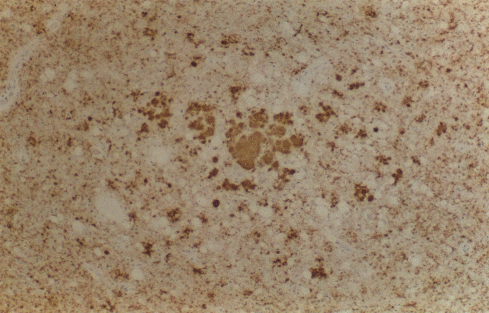 Figure 9C
Figure 9C
Immunocytochemistry for PrP in the thalamus shows several large multicentric plaques
(centre) with perivacuolar and synaptic deposition in
the surrounding neuropil ©
The UK Creutzfeldt-Jakob Disease Surveillance Unit/The Lancet
Gerstmann-Sträussler-Scheinker syndrome
Gerstmann-Sträussler-Scheinker syndrome (GSS) is a disease of man and has symptoms that are Kuru-like. It is a familial disease and is often regarded as a genetically transmitted subclass of CJD cases. This disease can be transmitted to laboratory animals.
Fatal Familial Insomnia
Fatal Familial Insomnia is a disease of man and results in progressive untreatable insomnia, loss of circadian rhythm, endocrine disorders, motor disorders, dementia. Again it is a familial (inherited) disease that can be transmitted to animals in the laboratory. In this form of the disease, it seems that hypothalamus function may be the initial target.
PRION PROTEIN
Preparations of highly purified infectious material contain large amounts PrP (figure 9 and 10). This is coded for by a host cellular gene and is a cell surface glycophospholipidylinositol (GPI)-anchored protein. Its normal function is not known. The infectious form has the same amino acid sequence and the same post-translational modifications as the normal form, but has a different conformation in diseased tissue. The normal form contains a lot of alpha-helix, whereas the disease-associated form contains a lot of beta-pleated sheet. The disease-associated form is known as PrPRES since it is more resistant to protease or as PrPSC since it was first found in scrapie infections.
Why is a protein infectious?
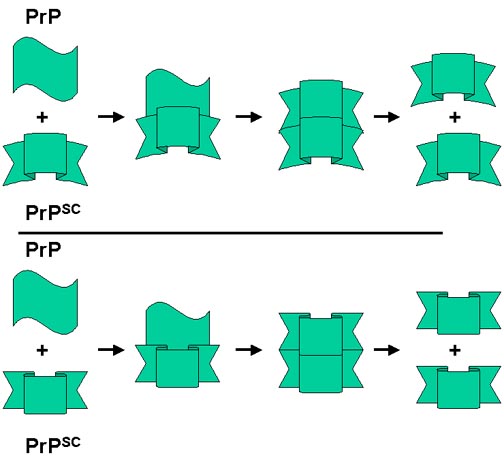
One hypothesis is that the resistant form can convert the normal form to the resistant form, which will then be able to convert more normal form to the resistant form; thus the rate of conversion will gradually amplify as the concentration of resistant form increases. At least some of this conversion appears to occur extracellularly.
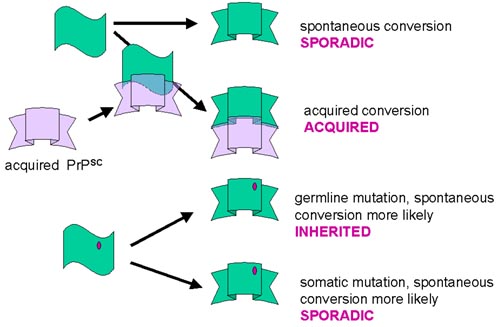
Acquired cases
Acquired cases may be due to being infected with the resistant form, which then may convert the person’s normal form to the PrPSC, and the process will gradually amplify as above.
Sporadic cases
Sporadic cases may be due to spontaneous conversion of normal to resistant form, and this process is then amplified as the resistant form recruits more normal form to the resistant form. Sporadic cases may also be due to somatic mutation, which makes the PrP more likely to undergo the spontaneous conversion to the resistant form, or may be acquired by an unknown mechanism.
Familial (hereditary) cases
In familial cases, mutations in the PrP gene have been observed. In the inherited form of this disease, the mutated form of the protein might have a greater likelihood of spontaneously changing to the resistant form and then the same recruitment process would occur. For at least some of these mutations, it appears that everyone who gets the mutant gene eventually develops CJD/GSS if they live long enough. The nature of the mutations in the inherited form can affect the clinical course of the disease.
IMMUNE RESPONSE
These unconventional viruses/agents do not cause an inflammatory response. They do not induce interferon. There is no antibody response against these agents. Hence it is not possible to screen people for exposure to these agents by looking for antibodies.
TREATMENT
To date, the prion diseases have been invariably fatal. Classic CJD usually results in death within a few months of the symptoms becoming obvious. The average time from symptoms becoming obvious to death in vCJD is longer - about sixteen months. Due to the poor prognosis for CJD patients, various drugs have been tested for efficacy but, so far, they seem to offer little if any positive effects, and if they were real they were very transient; moreover side effects of these drugs can be very serious.
Another approach has been to make antibodies which inhibit prion formation in mice. Part of the excitement here is because it seems that if one stops further PrPSC formation, cells can actually dispose of the PrPsc which has already formed. This approach has not yet been tried in humans.
One aspect to remember is that if there are drugs that slow progress, even if they do not cure someone with symptoms, they may be of use in the familial forms where they could be given before symptoms develop. This is a very new area in prion disease.
DIAGNOSIS
During life, a probable diagnosis is based on the clinical picture. EEG can provide useful supportive evidence in some cases. The wide range of symptoms and disease course make diagnosis difficult and prion diseases are often misdiagnosed. The final diagnosis is usually made from post-mortem examination of the brain. A brain biopsy can be used. Serology is of no use since the patient does not show an immunological response.
In the case of vCJD, MRI can be useful in diagnosis. A positive diagnosis can sometimes be made due to the presence of PrPSC in peripheral lymphoid tissue; for example, tonsil biopsies have been used. It is possible to make antibodies to PrPSC by using mice that have had the PrP gene deleted and are therefore not tolerized to the protein. These antibodies can then be used in a Western blot type of assay if enough prion material can be obtained from the patient.
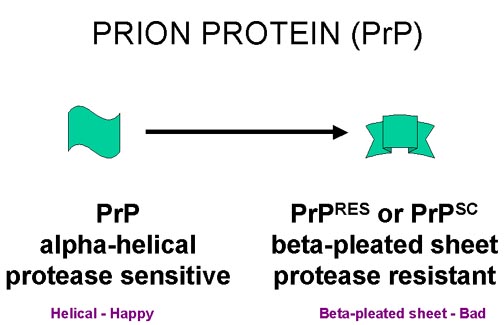 The prion protein PrP (encoded by a cellular gene
and made in normal cells) can exist in two forms. In diseased tissue the
protease-resistant form (PrPsc) with a lot of beta-pleated
sheet accumulates as 'amyloid plaques'
The prion protein PrP (encoded by a cellular gene
and made in normal cells) can exist in two forms. In diseased tissue the
protease-resistant form (PrPsc) with a lot of beta-pleated
sheet accumulates as 'amyloid plaques'Figure 10
Simplified model for prion disease.
PrPsc, the protease-resistant form of the molecule, acts as a 'template'. It associates with the helical form allowing the latter to be converted to the beta-pleated sheet resistant form (presumably by lowering the energy barriers that normally prevent this happening). There are now two molecules of the resistant form that can act as a template and so the process accelerates
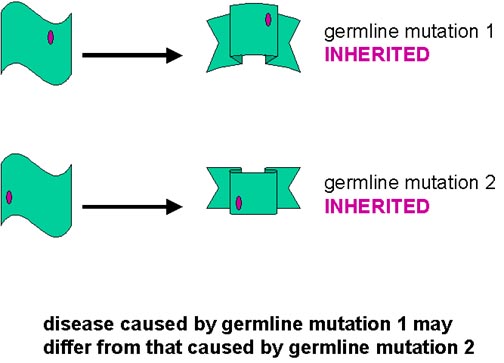
How can this model explain the sporadic, acquired or inherited form of the disease? The conversion from the alpha helical to the beta-sheet form may occur spontaneously, though very rarely (sporadic). The conversion may be catalyzed by PrPsc that comes from some exogenous source (acquired). Germ line mutations may make spontaneous conversion more likely (inherited). Somatic mutations may make spontaneous conversion more likely (sporadic). In this case, the mutant form could start the process of conversion and the resulting PrPsc molecules would then convert the normal form from surrounding cells
Why are there differences in prion diseases? There may be subtle differences in the protease-resistant form (PrPsc) of the prion protein according to the source of the PrPsc or the mutation involved. As indicated in the figure, the two related but subtly different forms of PrPsc convert the normal form to their own conformation. Thus, the final PrPsc product that accumulates depends on the form that initiated the process
This explanation may also be applied to the inherited forms. Different mutations may predispose the PrPsc to adopt slightly different protease-resistant forms spontaneously
TRANSMISSIBLE ENCEPHALOPATHIES AND
OTHER DISEASES
Amyloid plaques are seen in other CNS diseases - but the major components of amyloid plaques seen in, for example, Alzheimer's disease are NOT made of the same material as those seen in Kuru, CJD, GSS etc. Amyloid refers to the staining properties, and many glycosylated protein aggregates can have similar staining properties.
It is possible that the way in which prion diseases interfere with the function of cells in the CNS may pinpoint crucial processes in the CNS whose disturbance leads to progressive degeneration of nervous tissue. Understanding the nature of the pathogenesis of prions may help understanding of other CNS diseases.
Return to the Virology section of Microbiology and Immunology On-line
This page last changed on Wednesday, November 23, 2016
Page maintained by Richard Hunt