|
Dr Richard C. Hunt Medical Microbiology MBIM 650/720 |
VIROLOGY - CHAPTER SIX ONCOGENIC VIRUSES |
||||||||||||||||||||||||
|
TEACHING
OBJECTIVES To learn how cells become transformed by the virus To learn the differences between DNA and RNA tumor viruses To understand how RNA viral oncogenes result in cell transformation |
Cancers are the result of a disruption of the normal restraints on cellular proliferation. It is
apparent that the number of ways in which such a disruption can occur is strictly limited and
there may be as few as forty cellular genes in which disruption leads to unrestrained cell
growth. There are two basic classes of these genes in which mutation can lead to loss of
growth control: Viruses are involved in cancers because they can either carry a copy of one of these genes or can alter expression of the cell's copy of one of these genes. |
||||||||||||||||||||||||
|
To understand the discovery of cellular proto-oncogenes To learn how cellular oncogenes may cause cancer in the absence of a virus To learn understand how these discoveries led to the discovery of anti-oncogenes To understand how the discovery of anti-oncogenes showed how DNA viruses may cause cancer |
CLASSES OF TUMOR VIRUSES There are two classes of tumor viruses: the DNA tumor viruses and the RNA tumor viruses, the latter also being referred to as RETROVIRUSES. We shall see that these two classes have very different ways of reproducing themselves but they often have one aspect of their life cycle in common: the ability to integrate their own genome into that of the host cell. Such integration is not, however, a pre-requisite for tumor formation. If a virus takes up residence in a cell and alters the properties of that cell, the cell is said to be transformed. TRANSFORMATION BY A VIRUS MAY BE DEFINED AS: CHANGES IN THE BIOLOGIC FUNCTIONS OF A CELL THAT RESULT FROM REGULATION OF THE CELL BY VIRAL GENES AND THAT CONFER ON THE INFECTED CELL CERTAIN PROPERTIES CHARACTERISTIC OF NEOPLASIA. THESE CHANGES OFTEN (BUT NOT ALWAYS) RESULT FROM INTEGRATION OF THE VIRAL GENOME INTO THE HOST CELL GENOME Transformation often includes loss of growth control, ability to invade extracellular matrix and dedifferentiation. In carcinomas, many epithelial cells undergo an epithelial-mesenchymal transformation. Transformed cells often exhibit chromosomal aberrations. The region of the viral genome (DNA in DNA tumor-viruses or RNA in RNA-tumor viruses) that can cause a tumor is called an ONCOGENE. This foreign gene can be carried into a cell and cause it to take on new properties such as immortalization and anchorage-independent growth. The discovery of viral oncogenes in retroviruses led to the finding that they are not unique to viruses and homologous genes (called proto-oncogenes) are found in all cells. Indeed, it is likely that the virus picked up a cellular gene during its evolution and this gene has subsequently become altered. Normally, the cellular proto-oncogenes are not expressed in a quiescent cell since they are involved in growth (which is not occurring in most cells of the body) and development; or they are expressed at low levels. However, they may become aberrantly expressed when the cell is infected by tumor viruses that do not themselves carry a viral oncogene. We shall see later how this happens but it is clear that a virus may cause cancer in two ways: It may carry an oncogene into a cell or it may activate a cellular proto-oncogene. The discovery of cellular oncogenes opened the way to the elucidation of mechanisms by which non-virally induced cancers may be caused. We shall investigate what the protein products of the viral and cellular oncogenes do in the infected cell and in cells in which cellular proto-oncogenes are expressed. We shall see that their functions strongly suggest mechanisms by which cells may be transformed to a neoplastic phenotype. The discovery of cellular oncogenes led to the discovery of another class of cellular genes, the tumor repressor (suppressor) genes or anti-oncogenes. Initially, the involvement of viral and cellular oncogenes in tumors caused by retroviruses was much more apparent than the involvement of the DNA tumor virus oncogenes but the discovery of tumor repressor genes (as a result of our knowledge of how retroviruses cause cancer) led to the elucidation of the mode of action of DNA virus oncogenes. It should be noted that while viruses have been vitally instrumental in elucidation of the mechanisms of oncogenesis, most human cancers are probably not the result of a retroviral infection although retroviruses are important in cancers in some animals. |
||||||||||||||||||||||||
 The information flow in DNA tumor viruses is similar to that in eucaryotic
cells
The information flow in DNA tumor viruses is similar to that in eucaryotic
cellsFigure 1 |
|||||||||||||||||||||||||
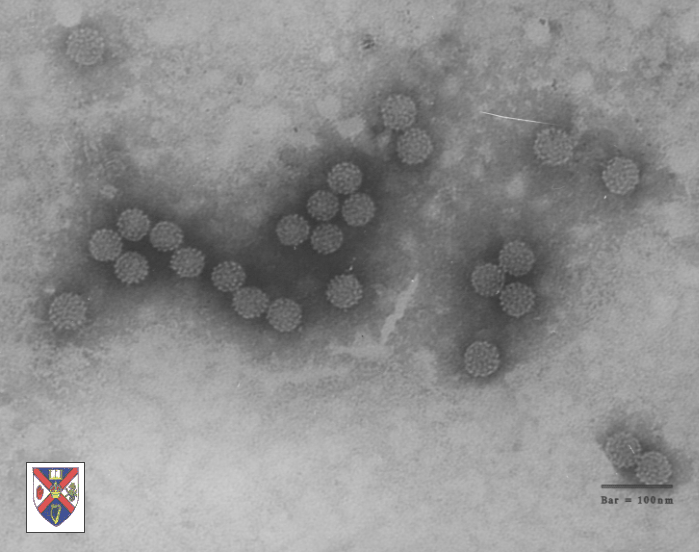 Papilloma virus Copyright 1994 Veterinary Sciences
Division, Queens University Belfast
Papilloma virus Copyright 1994 Veterinary Sciences
Division, Queens University Belfast |
DNA TUMOR VIRUSES
DNA tumor viruses have two life-styles: In permissive cells, all parts of the viral genome are expressed. This leads to viral replication, cell lysis and cell death In cells non-permissive for replication, viral DNA is integrated into the cell chromosomes (usually but not always) at random sites. Only part of the viral genome is expressed. The early, control functions (e.g. T antigens) of the virus, are expressed. Viral structural proteins are not expressed and no progeny virus is released. |
||||||||||||||||||||||||
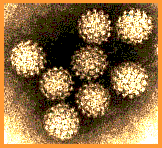 Papilloma virus Copyright Dr
Linda M Stannard, 1995
(used with permission)
Papilloma virus Copyright Dr
Linda M Stannard, 1995
(used with permission)
Figure 2 |
DNA TUMOR VIRUSES INVOLVED IN HUMAN CANCERS FAMILY: Papovaviridae - Papovaviruses 1) PAPILLOMAVIRUSES Papilloma viruses are wart-causing viruses that also certainly cause human neoplasms and cause natural cancers in animals. Warts are usually benign but can convert to malignant carcinomas. This occurs in patients with epidermodysplasia verruciformis (more here). Papilloma viruses are also found associated with human penile, uterine and cervical carcinomas and are very likely to be their cause; moreover, genital warts can convert to carcinomas. Squamous cell carcinomas of larynx, esophagus and lung appear very like cervical carcinoma histologically and these may also involve papilloma viruses. There are 51 types of papilloma viruses but, clearly, not all are associated with cancers; however, papillomas may cause 16% of female cancers worldwide and 10% of all cancers. |
||||||||||||||||||||||||
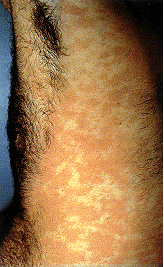 Epidermodysplasia verruciformis. This widespread, markedly
pruritic, erythematous eruption was eventually found to be caused by human papillomavirus infection.
International Association of Physicians in AIDS Care
Epidermodysplasia verruciformis. This widespread, markedly
pruritic, erythematous eruption was eventually found to be caused by human papillomavirus infection.
International Association of Physicians in AIDS Care
|
Vulvar, penile and cervical cancers associated with type 16 and type 18 papilloma viruses (and others) but the most common genital human papilloma viruses (HPV) are types 6 and 11. As might be expected if they are indeed the causes of certain cancers, types 16 and 18 cause transformation of human keratinocytes. In a German study, it was shown that 1 in 30 HPV type16-infected women will develop malignant disease while 1 in 500 infected people develop penile or vulvar cancer. Since not all infected persons develop a cancer, there are probably co-factors in stimulating the disease. Such co-factors have been identified in alimentary tract carcinomas in cattle where a diet containing bracken fern is associated with the disease. NOTE, HOWEVER: THE FACT THAT A VIRUS IS USUALLY FOUND IN ASSOCIATION WITH A NEOPLASM DOES NOT IN ANY WAY PROVE THAT THE TRANSFORMATION OF THE CELLS IS THE RESULT OF THE PRESENCE OF THE VIRUS. THE ASSOCIATION COULD BE CASUAL NOT CAUSAL. THE VITAL EXPERIMENT, DONE IN MANY ANIMAL SYSTEMS, WOULD BE TO INJECT THE VIRUS PURIFIED FROM A TUMOR INTO A HUMAN AND SEE IF THE TUMOR REDEVELOPS. FOR OBVIOUS REASONS THAT CRITICAL EXPERIMENT HAS NOT BEEN DONE. Nevertheless, the epidemiological data are very strong. 2) POLYOMA VIRUSES Simian virus 40 Polyoma virus Human polyoma viruses |
||||||||||||||||||||||||
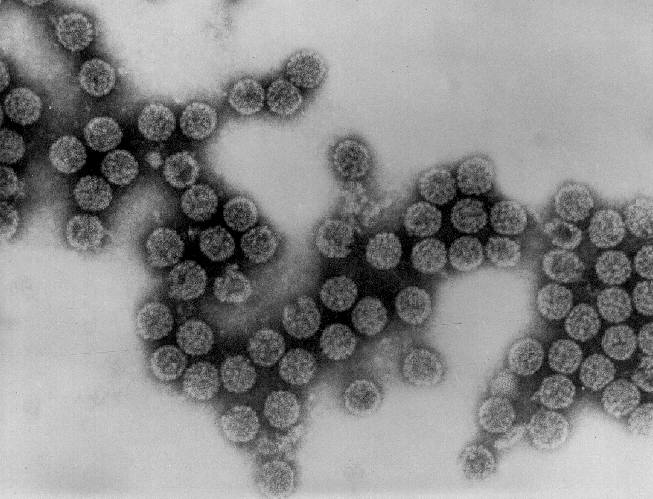 Transmission electron micrograph of polyomavirus SV40
Dr. Erskine Palmer CDC
Transmission electron micrograph of polyomavirus SV40
Dr. Erskine Palmer CDCFigure 4 |
Note: Polyoma viruses are usually lytic and when transformation occurs, it is because the transforming virus is defective. After integration into host DNA, only EARLY FUNCTIONS are transcribed into mRNA and expressed as a protein product. These are the TUMOR ANTIGENS. Because the expression of the genes for tumor antigens is essential for transformation of the cells, they may be classified as ONCOGENES. DEFINITION OF AN ONCOGENE: AN ONCOGENE IS A GENE THAT CODES FOR A PROTEIN THAT POTENTIALLY CAN TRANSFORM A NORMAL CELL INTO A MALIGNANT CELL. IT MAY BE TRANSMITTED BY A VIRUS IN WHICH CASE WE REFER TO IT AS A VIRAL ONCOGENE. SV 40 Tumor antigens are oncogenes
Two important points that should be noted about T antigens of DNA tumor viruses as oncogenes:
These properties should be contrasted with retroviral oncogenes to be discussed later |
||||||||||||||||||||||||
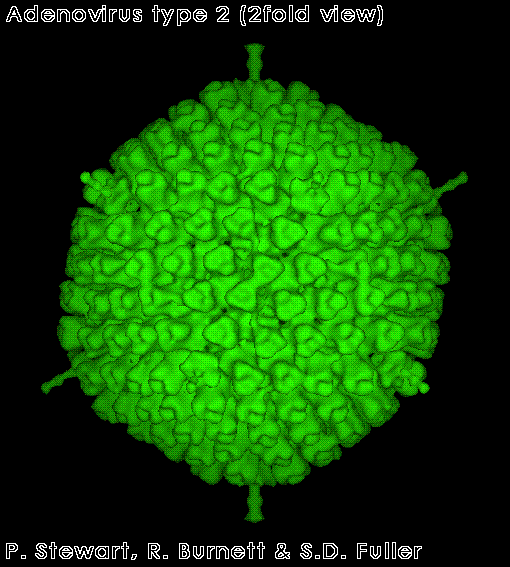 Adenovirus
Copyright
Dr Stephen
Fuller, 1998
Adenovirus
Copyright
Dr Stephen
Fuller, 1998 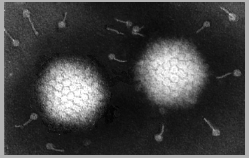 Adenovirus
Copyright Dr
Linda
M Stannard, University of Cape Town, South Africa, 1995
(used with permission). Adenovirus
Copyright Dr
Linda
M Stannard, University of Cape Town, South Africa, 1995
(used with permission).Figure 5 |
FAMILY: Adenoviridae ADENOVIRUSES These viruses are highly oncogenic in animals and only a portion of the virus is integrated into the host genome. This portion codes for early functions (E1A region contains the oncogenes that code for several T antigens). No humans cancers have been unequivocally associated with adenoviruses. E1A gene product (early non-structural protein) binds to the product of Rb gene (see below). Thus polyoma and adenoviruses seem to cause cell transformation in a similar manner: the integration of early function genes into the chromosome and the expression of these DNA synthesis-controlling genes without the production of viral structural proteins. |
||||||||||||||||||||||||
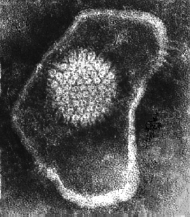 Herpes virus. Negative stain Copyright Dr
Linda M Stannard,
University of Cape Town, South Africa, 1995 (used
with permsssion).
Herpes virus. Negative stain Copyright Dr
Linda M Stannard,
University of Cape Town, South Africa, 1995 (used
with permsssion). 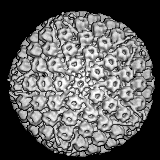 Liquid-Crystalline, Phage-like Packing of Encapsidated DNA in Herpes Simplex Virus
(F.P.Booy, W.W.Newcomb, B.L.Trus, J.C.Brown, T.S.Baker,
and A.C.Steven, in CELL, Vol 64 pp 1007-1015, March 8, 1991)
Liquid-Crystalline, Phage-like Packing of Encapsidated DNA in Herpes Simplex Virus
(F.P.Booy, W.W.Newcomb, B.L.Trus, J.C.Brown, T.S.Baker,
and A.C.Steven, in CELL, Vol 64 pp 1007-1015, March 8, 1991)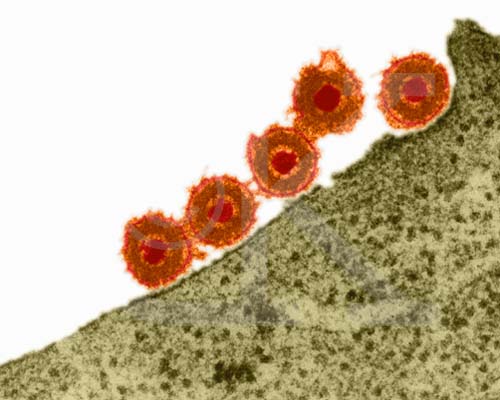 Herpes Simplex Virus (TEM x169,920)
Copyright Dr Dennis Kunkel (used with permission)
Herpes Simplex Virus (TEM x169,920)
Copyright Dr Dennis Kunkel (used with permission)Figure 6 |
FAMILY: Herpesviridae HERPESVIRUSES There is considerable circumstantial evidence that implicates these enveloped DNA viruses in human neoplasms. They are highly tumorigenic in animals. It is notable that herpes viruses exist primarily as episomes in the cell and do not integrate into the host cell genome. By the time that tumors arise, no trace of the virus can usually be found. Herpes virus DNA is found in only a small number of herpes-transformed cells. They may have a hit and run mechanism of oncogenesis, perhaps by causing chromosomal breakage or other damage. See below. Epstein-Barr virus This is the herpes virus that is most strongly associated with cancer. It is causally associated with:
|
||||||||||||||||||||||||
|
WEB RESOURCES Epstein–Barr virus infection: basis of malignancy and potential for therapy |
 Burkitt's Lymphoma caused by Epstein-Barr Virus The Johns Hopkins Autopsy Resource
(JHAR) Image Archive.
Burkitt's Lymphoma caused by Epstein-Barr Virus The Johns Hopkins Autopsy Resource
(JHAR) Image Archive. Figure 7 Epstein- Barr Virus |
A
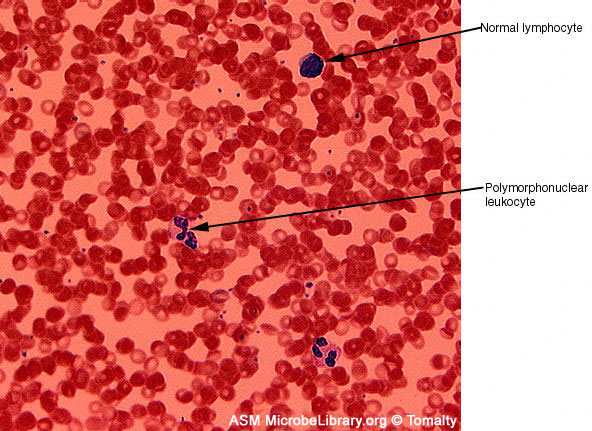 B
B 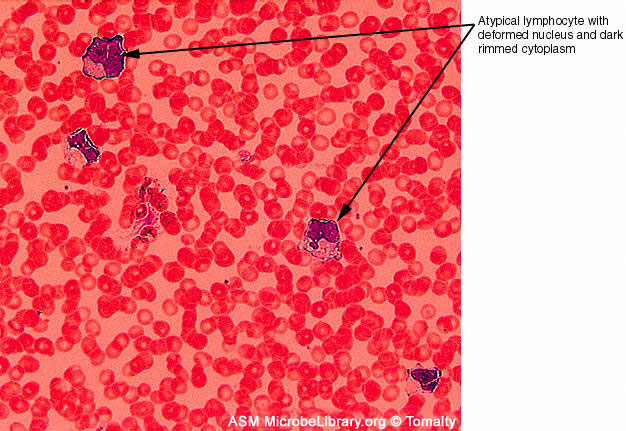 Peripheral blood smears from a healthy individual (A)
and a patient with infectious mononucleosis caused by Epstein-Barr virus
(EBV) (B). Both smears are stained with Giemsa stain ©
Gloria J. Delisle and Lewis Tomalty Queens University
Kingston, Ontario, Canada and The
MicrobeLibrary
Peripheral blood smears from a healthy individual (A)
and a patient with infectious mononucleosis caused by Epstein-Barr virus
(EBV) (B). Both smears are stained with Giemsa stain ©
Gloria J. Delisle and Lewis Tomalty Queens University
Kingston, Ontario, Canada and The
MicrobeLibrary
|
|||||||||||||||||||||||
|
Human cytomegalovirus This herpes virus is frequently associated with Kaposi's sarcoma but this is now thought probably to be caused by a newly-discovered herpes virus, human herpes virus 8. Herpes simplex II This virus was associated in epidemiological studies with cervical cancer. Now the evidence for papilloma virus being the causative agent of cervical cancer is far better For more on herpes viruses and the diseases that they cause, go to Virology Chapter 11 Herpes Viruses
|
|||||||||||||||||||||||||
|
FAMILY: Hepadnaviridae HEPATITIS B VIRUS |
|||||||||||||||||||||||||
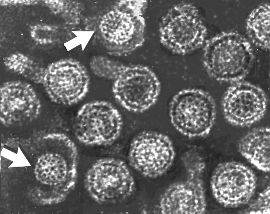 Hepatitis B virions: two exposed cores (indicated by
arrows)
Hepatitis B virions: two exposed cores (indicated by
arrows) |
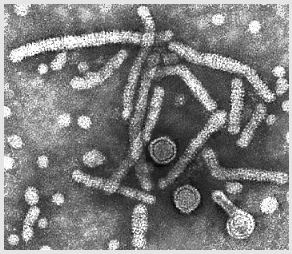 Hepatitis B virions
Hepatitis B virions |
 A diagrammatic representation of the hepatitis B virion and the surface antigen
components
A diagrammatic representation of the hepatitis B virion and the surface antigen
components |
|||||||||||||||||||||||
|
Figure 8 All four images: Copyright Dr Linda M Stannard, University of Cape Town, South Africa, 1995 (used with permission). |
|||||||||||||||||||||||||
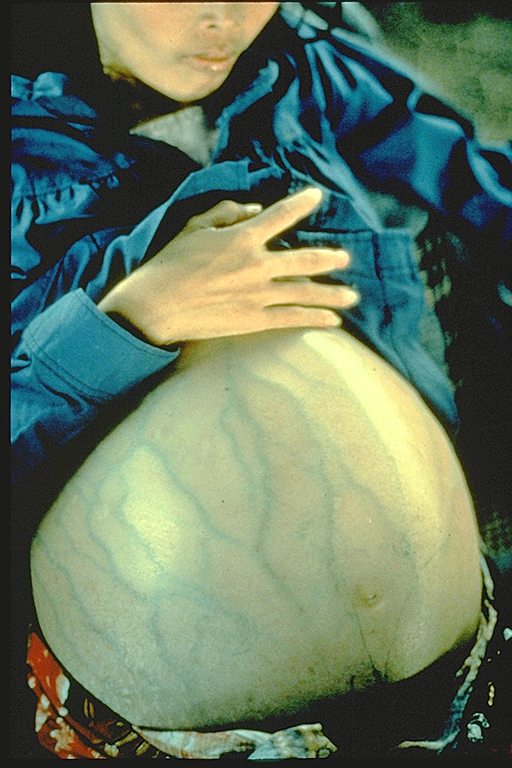 This woman has hepatitis B and is suffering from liver cancer. She was a Cambodian refugee
and died 4 months after she arrived in a refugee camp (average life expectancy after diagnosis of liver cancer is 6 months)
Immunization Action Coalition Courtesy of Patricia Walker, MD, Ramsey Clinic Associates, St. Paul, MN
This woman has hepatitis B and is suffering from liver cancer. She was a Cambodian refugee
and died 4 months after she arrived in a refugee camp (average life expectancy after diagnosis of liver cancer is 6 months)
Immunization Action Coalition Courtesy of Patricia Walker, MD, Ramsey Clinic Associates, St. Paul, MNFigure 9 |
Hepatitis B virus is very different from the other DNA tumor viruses.
Indeed, even though it is a DNA virus, it is much more similar to the
oncornaviruses (RNA tumor viruses) in its mode of replication. Hepatitis B is a vast public health
problem and hepatocellular carcinoma (HCC), which is one of world's most common cancers, may well be caused by
HBV. There is a very strong correlation between HBsAg (hepatitis B virus surface
antigen) chronic carriers and the incidence of HCC. In Taiwan, it
has been shown that HBsAg carriers have a risk of HCC
that is 217 times that of a non-carrier. 51% of deaths of HBsAg carriers are caused by
liver cirrhosis or
HCC compared to 2% of the general population.
NOTE: Hepatitis B virus is a DNA tumor virus BUT it has a very weird way of replicating itself. The DNA is transcribed into RNA not only for the manufacture of viral proteins but for genome replication. Genomic RNA is transcribed back into genomic DNA. This is called REVERSE TRANSCRIPTION. This is not typical of DNA tumor viruses but reverse transcription is a very important factor in the life cycles of RNA-tumor viruses. See below. |
||||||||||||||||||||||||
|
WEB RESOURCES |
|||||||||||||||||||||||||
 Human
immunodeficiency virus
Copyright Department of Microbiology, University of
Otaga, New
Zealand. Human
immunodeficiency virus
Copyright Department of Microbiology, University of
Otaga, New
Zealand.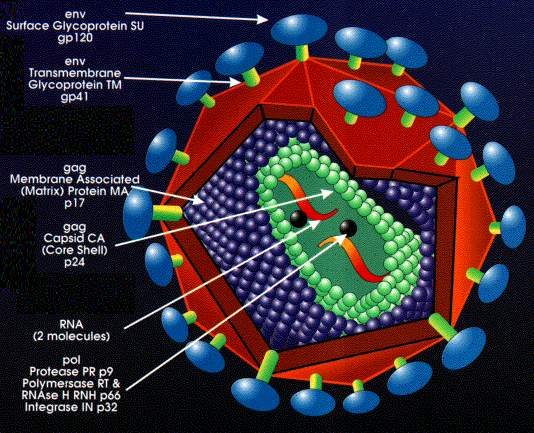 Structure of a retrovirus: (The virus shown is
human immunodeficiency virus-1) From the Harvard AIDS Institute Library of Images, courtesy of Critical
Path AIDS Project, Philadelphia.
Structure of a retrovirus: (The virus shown is
human immunodeficiency virus-1) From the Harvard AIDS Institute Library of Images, courtesy of Critical
Path AIDS Project, Philadelphia.Figure 10
|
RNA TUMOR VIRUSES
(RETROVIRUSES)
Retroviruses are different from DNA tumor viruses in that their genome is RNA but they are similar to many DNA tumor viruses in that the genome is integrated into host genome. Since RNA makes up the genome of the mature virus particle, it must be copied to DNA prior to integration into the host cell chromosome. This life style goes against the central dogma of molecular biology in which DNA is copied into RNA. Retrovirus structure The outer envelope comes from the host cell plasma membrane Coat proteins (surface antigens) are encoded by env (envelope) gene. One primary gene product is made but this is cleaved so that there are more than one surface glycoprotein in the mature virus (cleavage is by host enzyme in the Golgi apparatus). Inside the membrane is an icosahedral capsid containing proteins encoded by the gag gene (Group- specific AntiGen). Gag- encoded proteins also coat the genomic RNA. Again there is one primary gene product. This is cleaved by a virally-encoded proteins (from the pol gene) There are two molecules of genomic RNA per virus particle with a 5' cap and a 3' poly A sequence. Thus, the virus is diploid. The RNA is plus sense (same sense as mRNA). About 10 copies of reverse transcriptase are present within the mature virus, these are encoded by the pol gene. Pol gene codes for several functions (again, as with gag and env, a polyprotein is made that is then cut up) |
||||||||||||||||||||||||
 Structure of RSV protease bound to a peptide analog of the HIV cleavage
site
Structure of RSV protease bound to a peptide analog of the HIV cleavage
siteRequires Netscape and a Chime plug-in. Get Chime here - Click on thumbnail to open file Figure 12 |
The pol gene products are:
|
||||||||||||||||||||||||
 Human T-lymphocyte Virus Attacking a T-lymphocyte (TEM x26,400)
Copyright Dr Dennis Kunkel (used with permission)
Human T-lymphocyte Virus Attacking a T-lymphocyte (TEM x26,400)
Copyright Dr Dennis Kunkel (used with permission)Figure 13 |
GROUPS OF RETROVIRUSES 1) ONCOVIRINAE Viruses in this group that cause tumors in humans are: HTLV-1 (human T-cell lymphotropic virus) (Information Box): Causes Adult T-cell leukemia (Sezary T-cell leukemia) which is found in some Japanese islands, the Caribbean, Latin America (Information Box) and Africa. HTLV-1 is sexually transmitted (Information Box) HTLV-2: Hairy cell leukemia (Information Box) 2) LENTIVIRINAE 3) SPUMAVIRINAE
|
||||||||||||||||||||||||
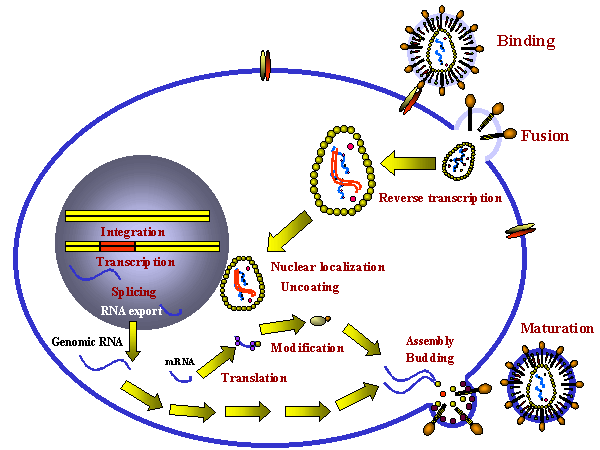 Stages in the productive infection of a cell by a retrovirus
Stages in the productive infection of a cell by a retrovirusFigure 14 |
INFECTION AND TRANSFORMATION OF A CELL BY A RETROVIRUS
The following stages occur in the infection process: 1) Binding to a specific cell surface receptor 2) Uptake by endocytosis or by direct fusion to the plasma membrane. The virus may need entry into a low pH endosome before fusion can occur although some (e.g. HIV) can fuse directly with the plasma membrane 3) RNA (plus sense) is copied by reverse transcriptase to minus sense DNA. Here, the polymerase is acting as an RNA-dependent DNA polymerase. Note: reverse transcriptase is a DNA polymerase and it therefore needs a primer. This is a tRNA that is incorporated into the virus particle. 4) RNA is displaced and degraded by a virus-encoded RNase H activity. Reverse transcriptase now acts as a DNA-dependent DNA polymerase and copies the new DNA into a double strand DNA. This is the provirus. 5) Double strand DNA is integrated into host cell DNA (see below) using a virally encoded integrase enzyme. This DNA is copied every time cellular DNA is copied. Thus, at this stage the provirus is just like a normal cellular gene. 6) Full length, genomic RNA (plus sense) is copied from integrated DNA by host RNA polymerase II. It is capped and poly adenylated
Note: mRNA comes from splicing genomic RNA or is the genomic RNA. As a result, both mRNA and genomic RNA must be the same sense - since mRNA must be plus sense, the genomic RNA of all retroviruses must also be plus sense. An advantage of this mode of replication is that it allows growth in terminally differentiated cells since the only host cell polymerase usurped by the virus is RNA polymerase II which is present in all cells. |
||||||||||||||||||||||||
|
MECHANISM OF VIRAL GENOME REPLICATION If host RNA polymerase II is used to copy the DNA back to RNA, there are major problems with having a DNA provirus form but an RNA genome in the mature virus particle These problems include:
Failure to copy the entire gene This means that either the DNA copy of the viral RNA genome virus must integrate into host DNA downstream from a host promotor and upstream from host termination sites (a tall order indeed!) or it must find a way of providing its own control sequences (which, as we said, are not copied into progeny genome). It does the latter in a most complex manner. |
|||||||||||||||||||||||||
| A B Figure 15 |
How can a retrovirus provide its own control promotors and enhancers if they are not
transcribed when the DNA provirus is copied to the genomic RNA form?
Here is a brief (and very incomplete) summary of how a retrovirus does it: 1) The viral RNA is composed of three regions. At each end are repeats (called, not surprisingly, terminal repeats). The repeat sequences (R) (shown in green) do not code for proteins. In between the two repeats, there is a unique (not repeated) region that contains the viral genes that code for the proteins (GAG, POL and ENV) plus other unique sequences at either end that do not code for protein. At the 5' end of the RNA genome is the U5 region and at the 3' end is the U3 region. PBS (in above diagram) is the primer binding site. The tRNA binds here when RT starts copying. PPT is a polypurine tract. |
||||||||||||||||||||||||
|
MOVIE LTR formation |
2) In the integrated form (when transcribed into DNA and inserted into the host cell chromosome), the provirus is more complicated. We find that part of the 3' unique region (called U3) of the RNA genome has been copied and transposed to the opposite end of the genome. Conversely, part of the 5' end of the unique region (called U5) has been copied and transposed to the other end. This gives the integrated DNA the structure shown in figure 15B. For convenience, only one strand of the DNA is shown. Now, of course, there are larger
terminal repeats since the U3 and U5 regions are also repeated. The U3-R-U5 regions
are known as long terminal repeats or LTRs. The U3 region contains all of
the promotor information that is necessary to start RNA transcription at the beginning
of the R (repeat) region while the U5 region contains all of the information necessary
to terminate after the other R repeat. In addition, the LTRs contain information that
enhances the degree of transcription of the three retroviral genes (enhancer regions).
These enhancers can be up or downstream from the protein-encoding part of
the genes. |
||||||||||||||||||||||||
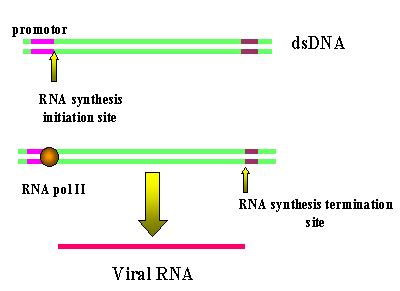 The transcription of a retrorviral DNA with LTRs by RNA
polymerase II results in the loss of the LTRs
The transcription of a retrorviral DNA with LTRs by RNA
polymerase II results in the loss of the LTRs Figure 16 Animated version here (requires IE5) |
Host RNA polymerase II copies the provirus DNA to genomic RNA which can be also spliced to mRNAs. Since the polymerase starts after the promotor (in U3), at the transcription initiation site, it begins exactly at the beginning of the R region (see diagram below). Thus we get a faithful (almost-see below) copy of the RNA that entered the cell. The termination sequences and poly A signal are in U5 which is also not copied. Note: Because of this mechanism, there can be only one promotor site (from U3) for all three viral genes so they must be all transcribed together. Splicing (enzymes from the host cell nuclear splicing machinery) gives the individual mRNAs where necessary. See notes on HIV in which this has been well elucidated. Unlike the situation with DNA tumor viruses, there is no distinction between early/late functions. Note: You may ask why, if U5 contains the termination and polyadenylation sites, does the transcript not just terminate after the first R region in the above diagram and never get into the structural genes. The termination site in the first U5 is suppressed, often by complex secondary structure mechanism. In some retroviruses there is a sequence in the gag gene that provides the context to suppress the termination activity of the first U5. Clearly the second U5 does not have a gag gene following it. Note: The copying of the RNA and the synthesis of the complementary DNA strand are carried out by reverse transcriptase. Reverse transcriptase is an RNA-dependent DNA polymerase and, like all DNA polymerases, it needs a primer. This is a cellular tRNA that is packaged within the viral particle. This strategy of virus replication in which viral RNA is first copied to DNA (by reverse transcriptase) which then gives rise to mRNA and protein poses a problem for the virus. The initial step (RNA to DNA) is carried out by a viral enzyme which is not normally in the cell. Yet this transcription step must occur before any mRNA transcription or protein translation can occur. The problem is solved by the virus carrying about 10 copies of the reverse transcriptase protein into the cell with it. These were packaged when the virus was assembled in the previous host cell.
|
||||||||||||||||||||||||
| ONCOGENES IN RETROVIRUSES | |||||||||||||||||||||||||
 Typical retrovirus structure and the structure of a retrovirus with an
oncogene (Rous Sarcoma Virus)
Typical retrovirus structure and the structure of a retrovirus with an
oncogene (Rous Sarcoma Virus)
Figure 17
|
The structure shown in figure 15A and the upper part of figure 17 is that of a typical retrovirus with three structural genes (gag, pol and env) but none of these is oncogenic. If the virus is to transform a cell, in addition to, or instead, of part of the gag/pol/env genome, it must have sequences that alter cellular DNA synthesis and provide the other functions that are typical of a transformed cell. Thus we also find an ONCOGENE (onc) in the viral genome of many retroviruses that transform cells to neoplasia (figure 17). Definition of virally-induced transformation: The changes in the biologic function and antigenic specificity of a cell that result from integration of viral genetic sequences into the cellular genome and that confer on the infected cell certain properties of neoplasia. Note, however, that transformation can be induced by factors other than viruses e.g. carcinogens. WHAT ARE THE ONCOGENIC GENES IN RETROVIRUS? In retroviruses, these were first discovered as an extra gene in Rous sarcoma
virus (RSV)
|
||||||||||||||||||||||||
 Some retroviruses that have an oncogene that
replaces their normal genes
Some retroviruses that have an oncogene that
replaces their normal genesFigure 18 |
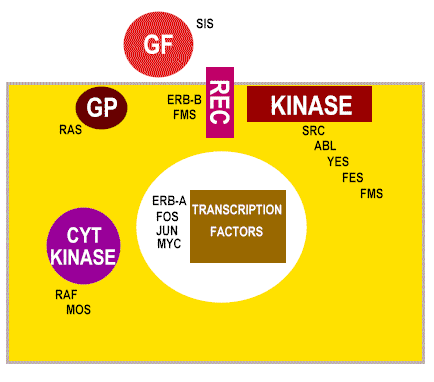
In sharp contrast to RSV, many retroviruses have lost part of their genome to accommodate an oncogene (Figure 18). This has two consequences: 1) The protein encoded by the oncogene is often part of a fusion protein with other virally-encoded amino acids attached 2) Virus is in trouble as cannot make all of itself. To replicate and bud from the host cell needs products of another virus, that is a helper virus About forty oncogenes have now been identified. Note that they are referred so by a three letter code (e.g. src, myc) often reflecting the virus from which they were first isolated. Some viruses can have more than one oncogene (e.g. erbA, erbB). Here are a few of the most studied:
|
||||||||||||||||||||||||
|
CELLS HAVE PROTO-ONCOGENES Once retroviral oncogenes had been discovered, a surprising observation was made: Unlike the situation with DNA virus oncogenes which are true viral genes, there are homologs of all retrovirus oncogenes in cells that are not infected by a retrovirus. These cellular homologs are often genes involved in growth control and development/differentiation (as might be expected) and have important non-transforming functions in the cell; some can cause cancer under certain circumstances and, presumably, those not shown to cause cancer have the ability to do so under the correct conditions. The cellular homologs of viral oncogenes are called proto-oncogenes. To distinguish viral oncogenes from cellular proto-oncogenes, they are often referred to as v-onc and c-onc respectively. Note: c-oncs are not identical to their corresponding v-oncs. It appears that the virus has picked up a cellular growth controlling or differentiation gene and, after the gene was acquired by the virus, it has been subject to mutation. Definition of a proto-oncogene: A host gene that is homologous to an oncogene that is found in a virus but which can induce transformation only after being altered (such as mutation or a change of context such as coming under the control of a highly active promotor). It usually encodes a protein that functions in DNA replication or growth control at some stage of the normal development of the organism. CHARACTERISTICS OF CELLULAR PROTO-ONCOGENES 1) These are typical cellular genes with typical control sequences. As with most eucaryotic genes, most have introns (retroviral oncogenes do not) 2) They show normal Mendelian inheritance 3) As with all genes in the eucaryotic genome, they are always at same place in genome (cf. what would be expected of endogenous retroviruses that had, over time, become incorporated into the cellular genome) 4) There are no LTR sequences (v-oncs always are in an LTR context) 5) Viral oncogenes are most like the c-onc of the animal from which the virus is thought to have acquired the gene. Thus, v-src of RSV is more like chicken src than human src. Note: v-onc was picked up accidentally by the virus from the genome of a previous host cell 6) Cellular oncogenes are expressed by the cell at some period in the life of the cell, often when the cell is growing, replicating and differentiating normally. They are usually proteins that are involved in growth control. 7) Cellular oncogenes are highly conserved If v-onc and c-onc are so alike, why does the viral oncogene (v-onc) introduced by a virus cause havoc in the cell? This is due to differences in the genes, mutations that have occurred in the gene once it was picked up by the virus. Such changes include: 1) Amino acid substitutions or deletions--gives different translation products 2) Many v-oncs are fusion proteins 3) V-oncs are inserted into the host genome along with LTRs which contain promotors/enhancers. This is likely to result in over expression of a gene that we know is probably involved in control of DNA transcription and replication!
|
|||||||||||||||||||||||||
CHRONICALLY TRANSFORMING RETROVIRUSES DO NOT HAVE V-ONC The observation that an acutely transforming virus such as RSV contains an extra gene, the oncogene, explains their high neoplastic potential but, in contrast, chronically transforming retroviruses only produce tumors slowly and they carry no gene equivalent to a v-onc. At best, these viruses have just the three usual viral genes (gag/pol/env). An example is avian leukosis virus (ALV). How do chronically transforming viruses induce a tumor if they do not have an oncogene? A seminal observation was made: Just as any other retrovirus does, ALV can integrate
into the cell genome at many different sites but, in ALV-induced tumors, the virus
is ALWAYS found in a similar position (very important!). This means that the
crucial transforming event must be rare and that the cells that form the tumor are a clone
(cf. the acute transformers which integrate all over the place). In all cases of
ALV-induced tumors, the viral genome is inserted near a cellular gene called c-myc.
This is the cellular proto-oncogene that, in an altered form (i.e. as a
v-onc), is
carried by some acutely transforming retroviruses (e.g. avian myelocytoma virus
which causes carcinoma, sarcomas and leukemias). In addition, the level of translation of
c-myc in the ALV-transformed cell is much greater than in uninfected cells. Thus,
inserting the genome of ALV and other chronically transforming retroviruses next to a
c-onc has the same effect as carrying in a v-onc. |
|||||||||||||||||||||||||
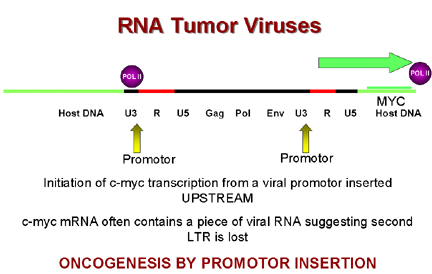 Oncogenesis by
promotor insertion
Oncogenesis by
promotor insertion Figure 19
|
So, in integration, the virus comes to lie upstream from c-myc which then comes under the influence of the strong LTR promotors of the virus which leads to over expression of c-myc. This is called oncogenesis by promotor insertion (Figure 19). But in some tumors the virus is downstream from the c-myc gene. However, we saw that LTRs also have enhancers in addition to promotors. We know that enhancer sequences can be upstream or downstream to have their effect. This is called oncogenesis by enhancer insertion (Figure 20). Why is insertion near c-myc important? The protein coded for by this gene is found in the nucleus of normal cells and is involved in control of DNA synthesis. It can be shown that over-expression of c-myc leads to rapid DNA replication.
|
||||||||||||||||||||||||
A
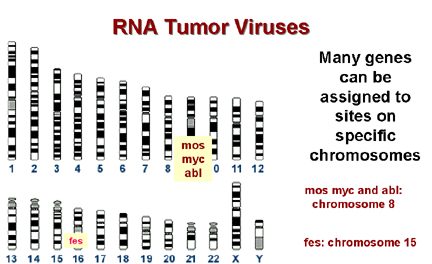 Many genes can be assigned to sites on specific chromosomes
Many genes can be assigned to sites on specific chromosomes
B |
CAN CELLULAR ONCOGENES BE INVOLVED IN NON-VIRALLY INDUCED CANCER?
Once it had been shown that viruses can either bring an oncogene into the cell or can take control of a cellular proto-oncogene to give rise to a tumor, the question arose of whether cellular proto-oncogenes could give rise to tumors in the absence of retroviral infection. The answer is yes! Other chromosomal rearrangements can bring a c-onc under the control of the wrong promotor/enhancer (Figure 21). Alternatively, the c-onc might be mutated in a particular way so that it was over-expressed or it might code for a mutant protein with an altered function.
Chromosomal mapping allows the precise localization of the site of a gene on a
particular chromosome and many cancers are associated with alterations in chromosomes,
particularly translocations (the breakage of a chromosome so that the two parts associate
with two parts of another chromosome) Many break sites in tumor cells are very close to a known c-onc. This is highly suggestive and unlikely to have occurred by chance!
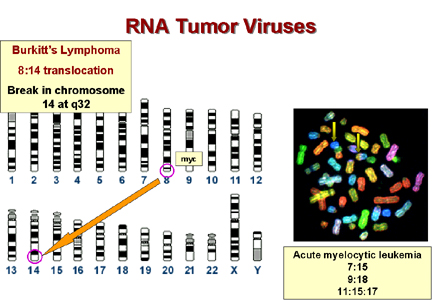
* In Burkitt's lymphoma the c-myc on chromosome 8 is brought to a site on chromosome 14 close to the gene for immunoglobulin heavy chains. It seems that the proto-oncogene may thus be brought under the control of the Ig promotor, which is presumably very active in B lymphocytes. This explains why this tumor arises in B cells. In other lymphomas, a c-onc is brought next to the immunoglobulin light chain promotor. These are also B cell lymphomas. Epstein-Barr virus is probably the cause of Burkitt's lymphoma. This is a herpes virus and herpes viruses commonly cause chromosomal breaks.
|
||||||||||||||||||||||||
|
IS THERE EVIDENCE THAT MUTATIONS IN CELLULAR ONCOGENES MIGHT ALSO RESULT IN TRANSFORMATION? The best evidence comes from the cellular oncogene that is the homologue of
the viral oncogene
found in the Harvey strain of murine sarcoma virus (the v-onc is called
HaRas). This c-onc was isolated from bladder carcinomas and compared to the normal
c-onc proto-oncogene. In many tumor cells only one change was found in the amino acid sequence
of the protein, glycine at amino acid position 12 was changed to valine. At position 12 only glycine and proline gave normal
growth. All other amino acids at this position gave a transformed cell. In a lung
carcinoma, the transforming DNA also contained c-HaRas, again it had a point mutation,
this time at position 61
|
|||||||||||||||||||||||||
|
WHAT IS THE NORMAL FUNCTION OF ONCOGENES? As mentioned above, c-oncs are normal cellular genes that are expressed
and function at some stage of the life of the cell. We should expect them to be
involved in DNA synthesis or perhaps the signaling pathways that lead to proliferation.
More than 40 oncogenes have been identified and there are probably a few
undiscovered ones We can sub-divide the cellular oncogenes into those that encode nuclear proteins and those that encode extra-nuclear proteins. The latter are mostly associated with the plasma membrane of the cell (Figure 22 and 23). Products of oncogenes that are nuclear proteins: e.g. myc, myb. These are involved in control of gene expression (that is the regulation of transcription-they are transcription factors) or the control of DNA replication. Neoplasia is associated with elevated transcription of the oncogene but strong expression is not always necessary, rather there is a need to make the gene constitutively active rather than under control of normal regulatory processes. Products of oncogenes that are cytoplasmic or membrane-associated proteins: e.g. abl, src, ras. This type does not exhibit altered expression but seems to convert from proto-oncogene to oncogene by mutation. Thus, in src-induced tumors, strong over expression of the oncogene has no effect.
|
|||||||||||||||||||||||||
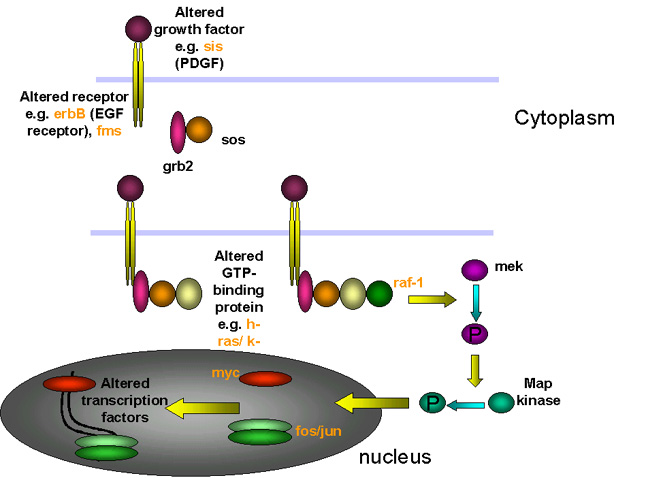 Ways in which altered proto- oncogenes might lead to cell
transformation
Ways in which altered proto- oncogenes might lead to cell
transformation Figure 22
|
In each of these cases, the mutation is dominant. Thus, for example if one allele of erb-B (a homolog of the EGF receptor) is mutated so that it is a constitutively switched on (i.e does not need epidermal growth factor to bind to switch on the tyrosine kinase activity), then the signal is on, regardless of the fact that the other allele is normal. |
||||||||||||||||||||||||
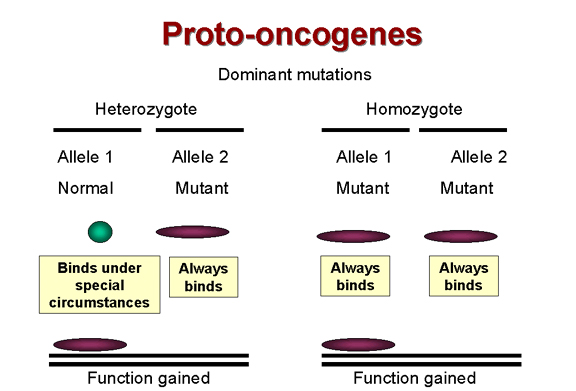 Dominant mutations are function
gained
Dominant mutations are function
gained Figure 24 |
ANTI-ONCOGENES (Tumor suppressor genes)The way in which retroviruses cause tumor formation via oncogenes was established before anything was known about how DNA tumor viruses cause tumors. Certainly, DNA tumor viruses carry oncogenes (e.g. SV40 T-antigen) but how do these proteins, encoded in true viral genes with no cellular homologs, cause the formation of tumors? It has long been known that most tumors are the result of dominant
mutations, i.e. a function is gained that makes the cell grow when it should
not
|
||||||||||||||||||||||||
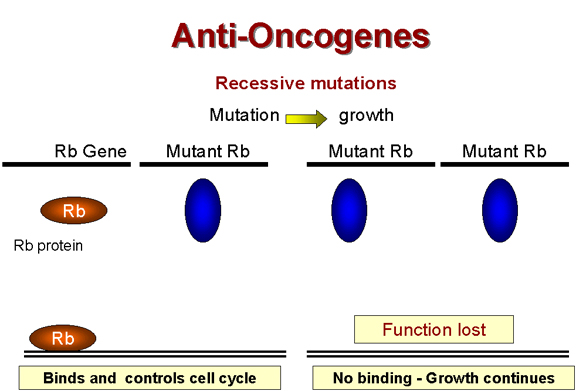 Recessive mutations are functions lost
Recessive mutations are functions lost Figure 24 |
Retinoblastoma: A recessive tumor There is a curious class of tumors that do not fit the usual characteristics in which the mutant oncogene is dominant over the wild type. In retinoblastoma, there appears to be a lesion that is recessive, that is
the cancer causing mutation causes a loss of function A heterozygote at the Rb allele still has normal Rb and tumors can still be suppressed but homozygote has no functional Rb and tumors cannot be suppressed
|
||||||||||||||||||||||||
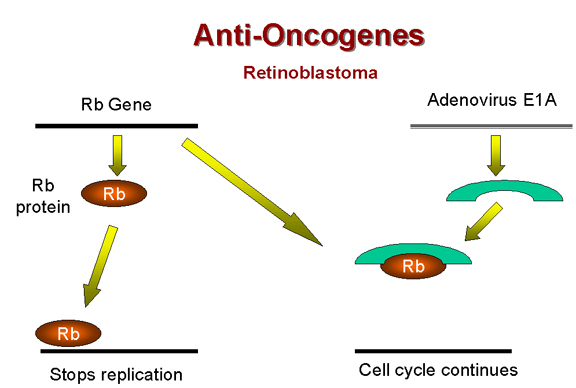 Rb and adenovirus
E1A
Rb and adenovirus
E1A Figure 25 |
Above, we have noted that the adenovirus E1A (early function)
protein is somehow involved in tumorigenesis. It has been found that E1A protein in the
transformed adenovirus-infected cell is complexed with a 105kD protein! This turns out to
be the Rb gene product (Figure 25). Thus, it seems that adenovirus may cause a cell to grow abnormally
by complexing (and thereby inactivating) a cellular protein whose normal function is
growth inhibition. Tumors caused by the inactivation of Rb gene product are, however,
rather rare |
||||||||||||||||||||||||
|
p53 and human cancer Over the past two decades, since its discovery in 1979, a gene known as the p53 gene (after the size of its encoded protein) has been linked to many cancers including many that are inherited. In these inherited cancers, it turns out that the p53 gene is mutant. Alterations in this protein seem to be the basis (direct or indirect) of most human cancers. In total, 60% of human cancers involve p53
|
|||||||||||||||||||||||||
|
80% of colon cancers involve the p53 gene Initially, it was thought that the p53 gene product caused cancers but further investigation showed the opposite, p53 is, like the retinoblastoma gene product, a tumor suppressor. p53 protein has been referred to as The Guardian of the Genome since it regulates multiple components of the DNA damage control system. How does p53 work in a functional cell? Normally, there are only a few of the suppressor p53 molecules in a healthy cell and these are constantly turning over; but when the DNA becomes damaged (perhaps by radiation or chemical mutagens) and DNA replication results, p53 turnover ceases. The rise in p53 stops DNA replication. p53 is a transcription factor. When it builds up, p53 binds to a specific site(s) on the chromosomes and switches on other genes and these, in turn, shut down mitosis. p53 can also act in another way, when it builds up it can set the cell on course to apoptosis. Whether or not p53 causes reversible growth arrest or apoptosis depends on the state of cellular activation; for example, extensive, unrepaired DNA damage can lead to sustained p53 production committing the cell to apoptosis. In inherited cancers, there is a mutation in the p53 gene; often it is a single point mutation and the protein can no longer bind to its correct site on the DNA and so cannot suppress DNA replication. Like the Rb gene, product, you would expect the effects of p53 to be recessive since the second normal p53 allele should make functional protein and should shut off DNA replication as usual; that is, if you are heterozygous for the mutation -- although, of course, you will only be one mutation away from carcinogenesis. So why do cells that are heterozygous for the p53 mutation also have problems? Unfortunately, p53 protein forms tetramers in a ribbon-like array and so if half of the p53 proteins are mutant, there is a good chance that each tetramer will have one mutant p53 molecule and this inactivates the tetramer, a dominant-negative effect.
|
|||||||||||||||||||||||||
 p53, hepatitis C
and papilloma virus
p53, hepatitis C
and papilloma virus Figure 26 |
Although we have learned a lot from families that inherit p53 mutations, it is clear that most p53 mutations come from non-inherited environmental factors: carcinogens (benzopyrene in smoke, aflatoxin in molds on peanuts and corn, UV light) that result in point mutations. Note: there are also gain of function p53 mutations that lead to very aggressive tumors. These turn on DNA replication genes. What has this got to do with DNA tumor viruses? Just as with retinoblastoma gene product, the presence of a virus can mimic mutation and take the tumor suppressor out of action by complexing it in an inactive form that cannot bind to the specific site on DNA. This is what appears to happen in hepatitis C which causes hepatocellular carcinoma. In the case of a human papilloma virus-infected cell, p53 is directed to a protease that recognizes a cleavage site in p53, thereby destroying it (Figure 26). Note: In radiation therapy, it was once thought that radiation damaged the DNA of dividing cells so that they could no longer divide. But in fact, the radiation only damages the DNA a bit which would not kill the cell but the bit of damage is enough to up-regulate the production of p53. Much research is now going on to see whether one can introduce healthy p53 genes into cells to shut down tumor growth. Thus, our knowledge of how retroviruses cause cancer has led to an understanding of the formerly cryptic manner in which DNA tumor viruses do the same thing. |
||||||||||||||||||||||||
|
This page copyright
2002, The
Board of Trustees of the University of South Carolina |
|||||||||||||||||||||||||